登陆查看更多优秀资源帖,与同道便捷交流讨论
您需要 登录 才可以下载或查看,没有帐号?立即注册
x
副作用限制临床应用
opiate 类药物是治疗急性和慢性疼痛的最有效药物。分别作为具有代表性的 opiate 类生物碱和合成 opiate 类药物,Morphine 和 Fentanyl 被用于癌痛治疗、麻醉镇痛、预防性镇痛以及术后多模式镇痛。虽然 opiate 类药物是有效的止痛药,但它们会导致严重的副作用,如呼吸抑制 (呼吸抑制所导致的死亡引发了广泛传播的 “ opiate 类药物危机”[1,2],尤其是在北美)、成瘾和便秘,从而限制了它们的临床应用。根据 2019 年发布的报告,超过 70% 的“ opiate 类药物危机”死亡是合成 opiate 类药物 (主要是Fentanyl 及其衍生物) 过量使用所致。这些副作用限制了 opiate 类药物在临床的应用。μOR 同为镇痛和副作用的主要受体
opiate 类药物的功能由四种 G 蛋白偶联受体 (GPCRs) 家族介导,即 μ、κ、δ 和伤害感受肽受体 (NOPR),在这些 opiate 受体中,μ 型 opiate 受体 (μOR) 被发现是镇痛和副作用的主要受体。有研究指出, opiate 样物质诱导的镇痛作用归因于 μOR 的 Gi 蛋白信号转导 (图 1),然而,其副作用 (呼吸抑制等) 究竟是由哪个信号通路产生,目前存在争议:一种观点认为由 β-arrestin 信号转导引起,另一观点认为与 G 蛋白门控的内向整流钾通道 (GIRK) 的信号转导有关。
图 1. Fentanyl 和Morphine 诱导的 μOR 信号传递和潜在的药理作用[1]
如何减少副作用?
今年 11 月,Cell 在线发表了题为 Molecular recognition of morphine and fentanyl by the human μ-opioid receptor 的研究性论文。这篇文章阐述了Fentanyl、Morphine 和其他 μOR 激动剂与 μOR 的结合方式,并揭示了它们与受体结合的关键差异。该研究还揭示了Morphine 和 Fentanyl 对 μOR 的 β-arrestin 蛋白活性起重要作用的结构因素,并为设计有效的、可能更安全的镇痛剂提供了结构模板[1]。
■ Morphine 和 Fentanyl 如何进行信号转导?
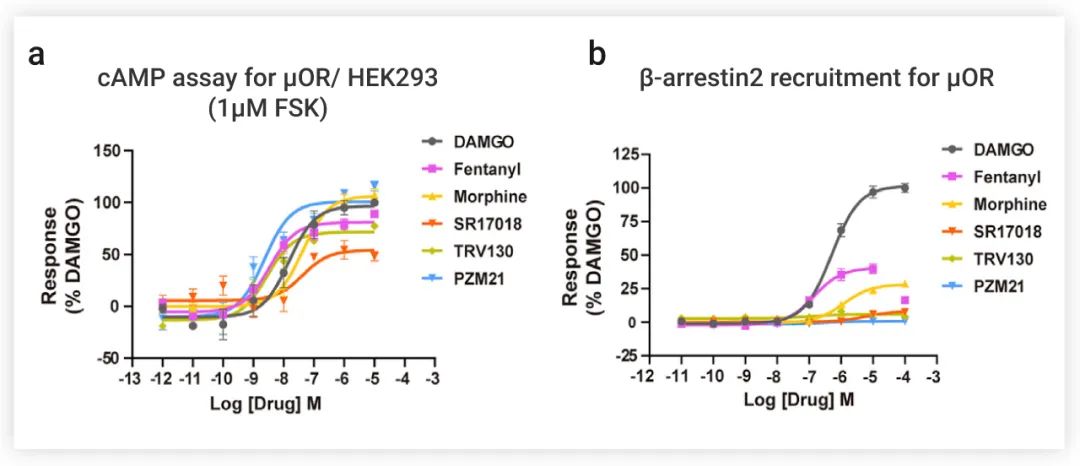
该研究首先表征了研究中使用的六种具有不同化学骨架的 opiate 类药物——Fentanyl 、Morphine 、SR17018、TRV130、PZM21 和 DAMGO (一种拟肽激动剂,可激活 μOR-Gi 复合物,作为对照)——的信号转导情况。
如果所示 (图 2a),所有五个配体都能激活 μOR,以抑制 cAMP 的产生,Fentanyl 、Morphine 和 PZM21 是完全激动剂,而 SR17018 和 TRV130 是部分激动剂。
在 β-arrestin 招募测定中没有检测到 PZM21 的反应信号,而对 SR17018 和 TRV130 只产生微弱的信号。相比之下,Morphine 和 Fentanyl 都能诱导强大的 β-arrestin 招募 (图 2b)。总而言之,Morphine 和 Fentanyl 不仅能完全激活 μOR,还可诱导强大的 β-arrestin 招募。
图 2. opiate 类激动剂的 cAMP 积累和 β-arrestin 招募实验[1]
a-b: opiate 类激动剂的 cAMP 积累 (a) 和 β-arrestin2 (b) 招募的剂量依赖反应曲线。DAMGO 为对照物,数据根据 DAMGO 的最大反应进行归一化。
■ 激活 μOR 的关键因素是什么?
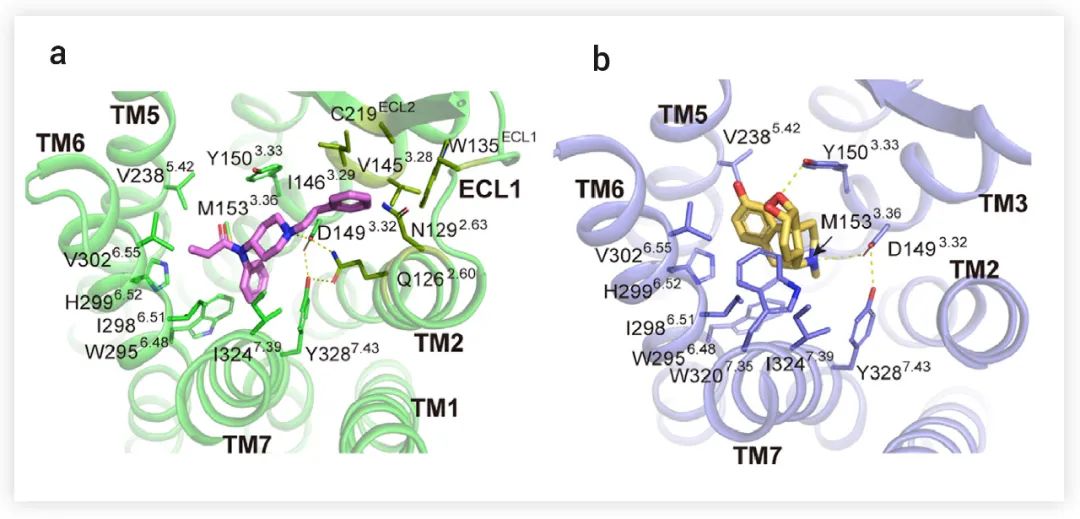
Morphine 和 Fentanyl 的化学骨架彼此不同,因此,作者分别研究了它们与 μOR 的特异性结合方式。研究表明,Fentanyl 分子在正位口袋中占据 "Y" 形构象,主要与跨膜结构域 (TMD) 的 TM2、TM3、TM6 和 TM7 的残基接触;Morphine 采用椭圆的 "O" 型结构,与来自 TM3、TM6 和 TM7 的疏水残基相互作用 (图 3)。该研究发现,D1493.32 与附近的残基 Q1262.60 和 Y3287.43(DQY 极性模体,以下简称 DQY模体)在两个结构中形成极性相互作用 (图 3a-b),并且 DQY 极性模体的突变,在很大程度上降低了Morphine 和 Fentanyl 的 G 蛋白和 β-arrestin 信号转导的效力,这表明 DQY 极性模体对 μOR 的激活至关重要。
图 3. Fentanyl (a) 和Morphine (b) 与 μOR 的相互作用[1]
在Morphine 和 Fentanyl 结合的 μOR 结构中,受体部分分别用绿色和蓝色标示。Minor pocket 中的残基用深绿色表示。Fentanyl :紫色;Morphine :金色。
此外,Morphine 和 Fentanyl 配体结合袋周围的残基突变,包括 Y1503.33、M1533.36、V2385.42、I2986.51 和 H2996.52,导致两种配体对 G 蛋白和 β-arrestin 信号的活性减弱 (图 4a-b)。
图 4. a: Morphine 和 Fentanyl 对 μOR 突变体的 Gi 激活减少所诱导的 cAMP 积累。每一栏的数值表示代表性的 μOR 突变体相对于 WT μOR的效力 (ΔpEC50) 的差异。b: Fentanyl (上) 和Morphine (下) 激活的小袋中 WT 和突变体 μORs 的 cAMP 测定[1]
■ 引起 β-arrestin 信号转导的关键因素又是什么?
作者对 μOR 正位结合口袋的残基进行了突变,并测试了这些突变体分别通过Fentanyl 、Morphine 和 DAMGO 激活 G 蛋白信号和 β-arrestin 招募的能力。结果显示,与 TM2 和 TM3 侧的残基相比,TM6 和 TM7 附近的残基突变对 β-arrestin 信号的影响更为明显。比如,来自 TM6 的 W295A 和来自 TM7 的W320A 突变在 cAMP 积累和 G 蛋白招募方面对 G 蛋白信号转导只有最小或部分影响,而它们几乎取消了Fentanyl 、Morphine 和 DAMGO 诱导的 β-arrestin 招募 (图 5b)。这说明,配体与 TM6/7 的相互作用对引起 β-arrestin 的信号转导至关重要。
图 5. TM6/7 的代表性残基的突变适度影响了 cAMP 反应,但取消了Fentanyl (a)、Morphine (b) 和DAMGO (c) 诱导的 β-arrestin 2 的招募[1]
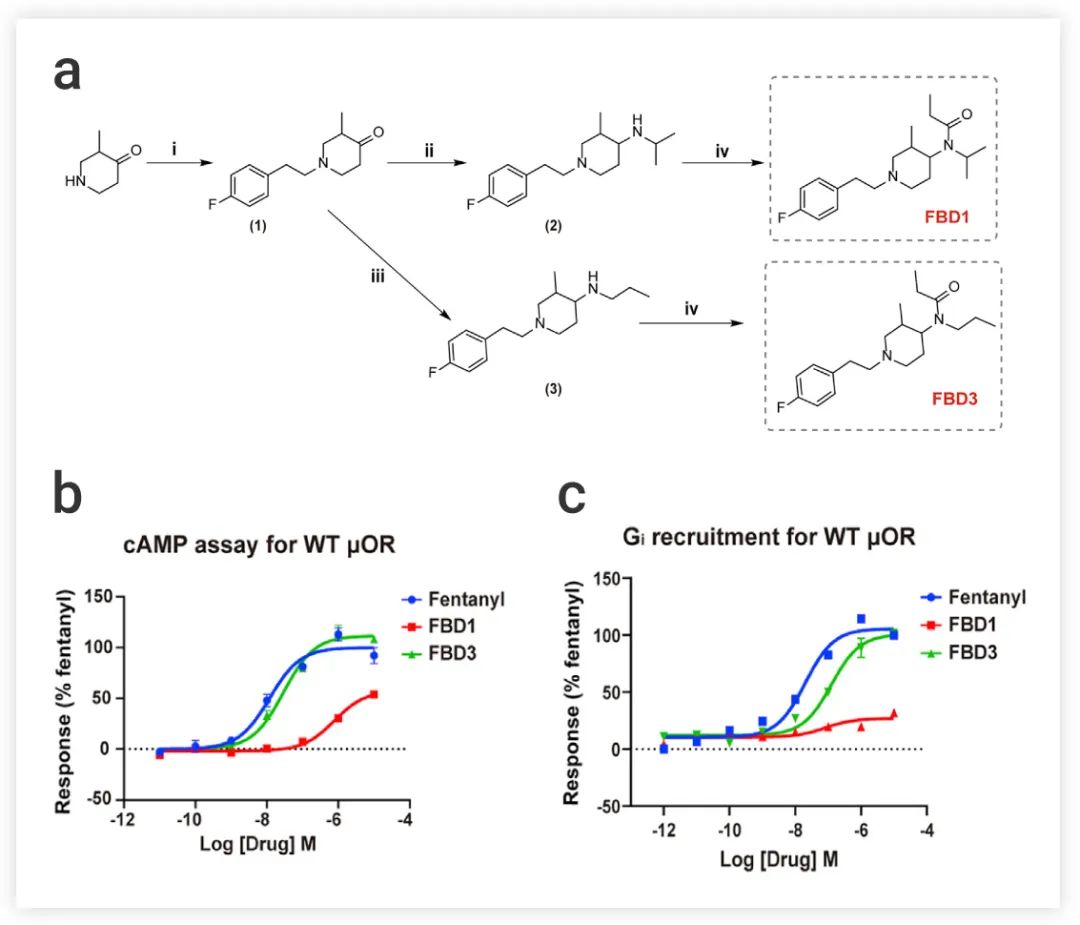
■ 设计分子降低 β-arrestin 活性了吗?
为了进一步验证上述发现,作者设计了两个结构相似的Fentanyl 衍生物 FBD1 (部分激动剂) 和 FBD3 (完全激动剂),以获得 β-arrestin 信号传导减少/消失但 G 蛋白活性相对完整的 μOR 激动剂 (图 6a),也就是说,FBD1 和 FBD3 与μOR 的 TM6/7 之间的相互作用被削弱。实验结果显示:与Fentanyl 相比,FBD1 和 FBD3 都显示出 β-arrestin 招募活性的极大降低,并且,FBD3 在 cAMP 抑制或 Gi 招募试验中显示出与Fentanyl 几乎相同的效力和效率 (图 6b-c)。
图 6. (a) Fentanyl 衍生物 FBD1 和 FBD3 的合成。(b-c) 以Fentanyl 为对照物 (b) FBD1 和 FBD3 的cAMP 积累试验和 Gi 招募 (c) 的剂量反应曲线图[1]
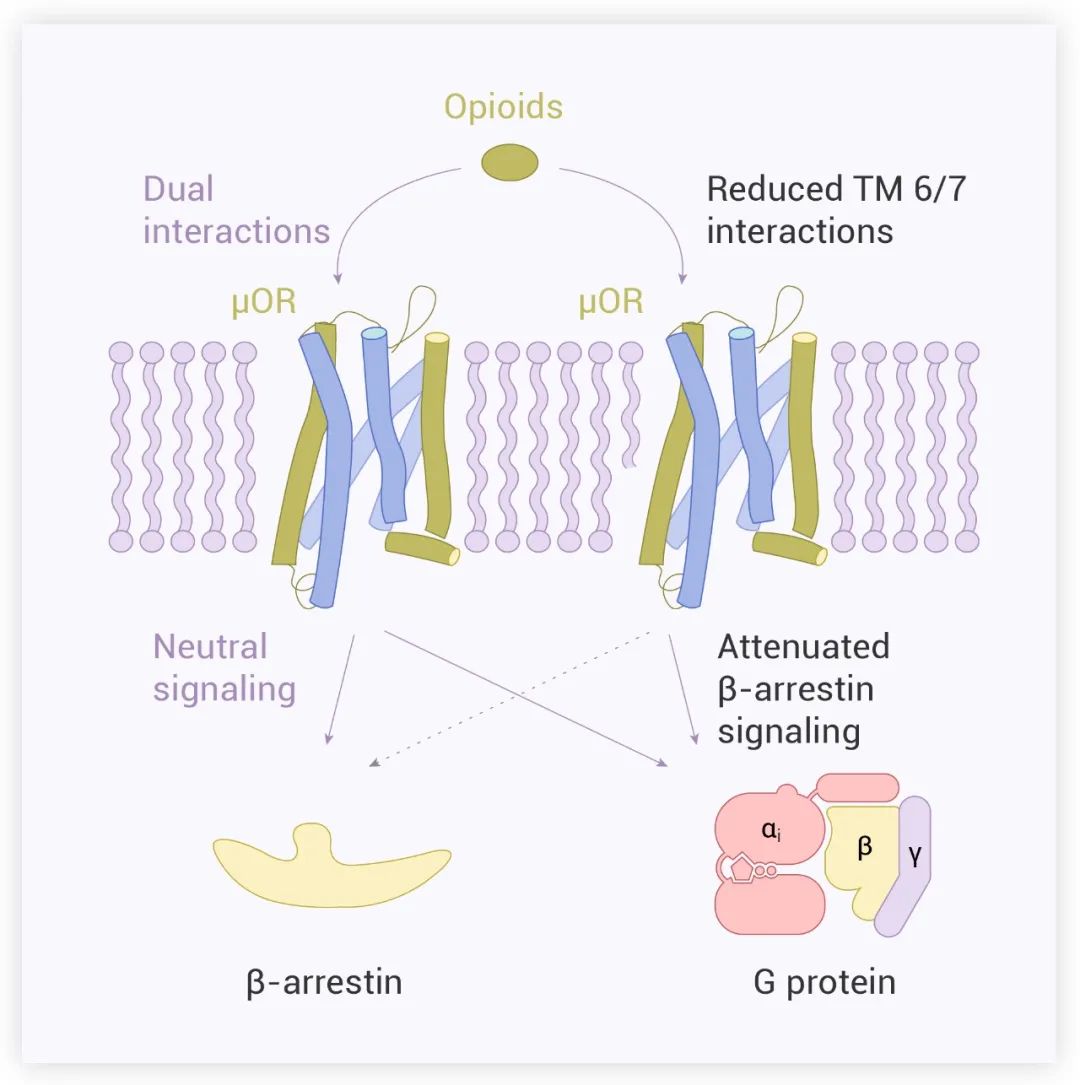
该研究进一步说明了配体与 TM6/7 的相互作用对引起 μOR 的 β-arrestin 信号转导至关重要,减少这种相互作用可能导致配体优先通过 G 蛋白途径转导信号 (图 7)。
图 7. μOR 的配体诱导的不同信号模型[1]
小结
这篇文章揭示了人类 μ 型 opiate 受体对Morphine 和 Fentanyl 的分子识别方式,并提出了基于Fentanyl 结构的类似物设计,削弱 μOR 的 β-arrestin 活性的思路。该研究有助于合理设计下一代镇痛剂,并有望在减少 opiate 类药物副作用的同时不影响其镇痛作用,甚至增强镇痛效果。
[size=14.6667px]
MCE 的所有产品仅用作科学研究或药证申报,我们不为任何个人用途提供产品和服务
参考文献
[1] Zhuang Y, H. Eric Xu, Xin Xie, Ming-Wei Wan, et al. Molecular recognition of morphine and fentanyl by the human μ-opioid receptor. Cell. 2022 Nov 10;185(23):4361-4375.e19.
[2] Vadivelu N, et al. The Opioid Crisis: a Comprehensive Overview. Curr Pain Headache Rep. 2018 Feb 23;22(3):16.
| 




 楼主
楼主