登陆查看更多优秀资源帖,与同道便捷交流讨论
您需要 登录 才可以下载或查看,没有帐号?立即注册
x
关于细胞的程序性死亡的相关研究一直是生命科学的热门领域,无论是持续火热的"铁死亡" (推文:铁死亡是什么,如何检测?您要的 “一文通” 来了)。还是正在生信领域大方异彩的 “铜死亡” (推文:空降 "热搜" 铜死亡丨解锁细胞死亡新方式),都涉及到 "离子转运"。在转运过程中,溶质载体 (Solute Carriers, SLC) 转运蛋白家族作为重要的膜转运蛋白家族,它们对葡萄糖、氨基酸和金属离子的转运有着重要的影响 (图 1)[1]。图 1. SLC 蛋白家族对不同离子以及氨基酸等转运[1]
SLC7A11----死亡调控途径中的双刃剑
SLC 家族成员 SLC7A11 转运蛋白在维持细胞内谷胱甘肽水平和保护细胞免受氧化应激诱导的细胞死亡方面具有重要作用,具有公认的促生存作用[3]。但有研究表明,胶质母细胞瘤细胞在葡萄糖剥夺条件下,通过 System Xc- (其中 SLC7A11 为催化亚基) 摄取胱氨酸会迅速诱导 NADPH 耗竭、活性氧物质积累和细胞死亡。 2020 年甘波谊团队在 Nature Cell Biology 发表的文章也表明,葡萄糖饥饿条件下,高 SLC7A11 表达反而促进细胞死亡,SLC7A11 就像是调节细胞氧化还原平衡和细胞死亡/存活方面的双刃剑[4]。
图 2. 葡萄糖饥饿条件下,高 SLC7A11 表达反而促进细胞死亡 [5] SLC7A11 和 SLC3A2 组成的胱氨酸/谷氨酸逆向转运蛋白 System Xc -,将胞内谷氨酸转出以 1:1 的比例来换取胞外的胱氨酸 (Cys2)
■ SLC7A11 高表达为何会 “促” 死亡?
这是因为胱氨酸是种不溶性氨基酸,为了防止细胞内高度不溶性胱氨酸的毒性积聚, SLC7A11high 细胞需要迅速将胱氨酸还原为半胱氨酸,而这个过程需要从葡萄糖-戊糖磷酸途径 (PPP) 获得大量 NADPH,这会对细胞NADPH 库会造成大量消耗,并使此类细胞产生葡萄糖和戊糖磷酸途径 (PPP) 依赖性 (如图 2) [4,5]。因此,当葡萄糖供应限制,氧化还原力不足, SLC7A11high 细胞内的胱氨酸或其他二硫化物分子的异常积累,诱发二硫化物应激触发细胞死亡。 今年 2 月,甘波谊团队发表的 Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis 一文更加详细阐明了这一死亡机制,研究表明肌动蛋白细胞骨架对二硫化应激的敏感性介导了双硫死亡 (disulfidptosis),并提出了在癌症治疗中靶向二硫化的治疗策略。
SLC7A11-high 细胞的独特细胞死亡方式
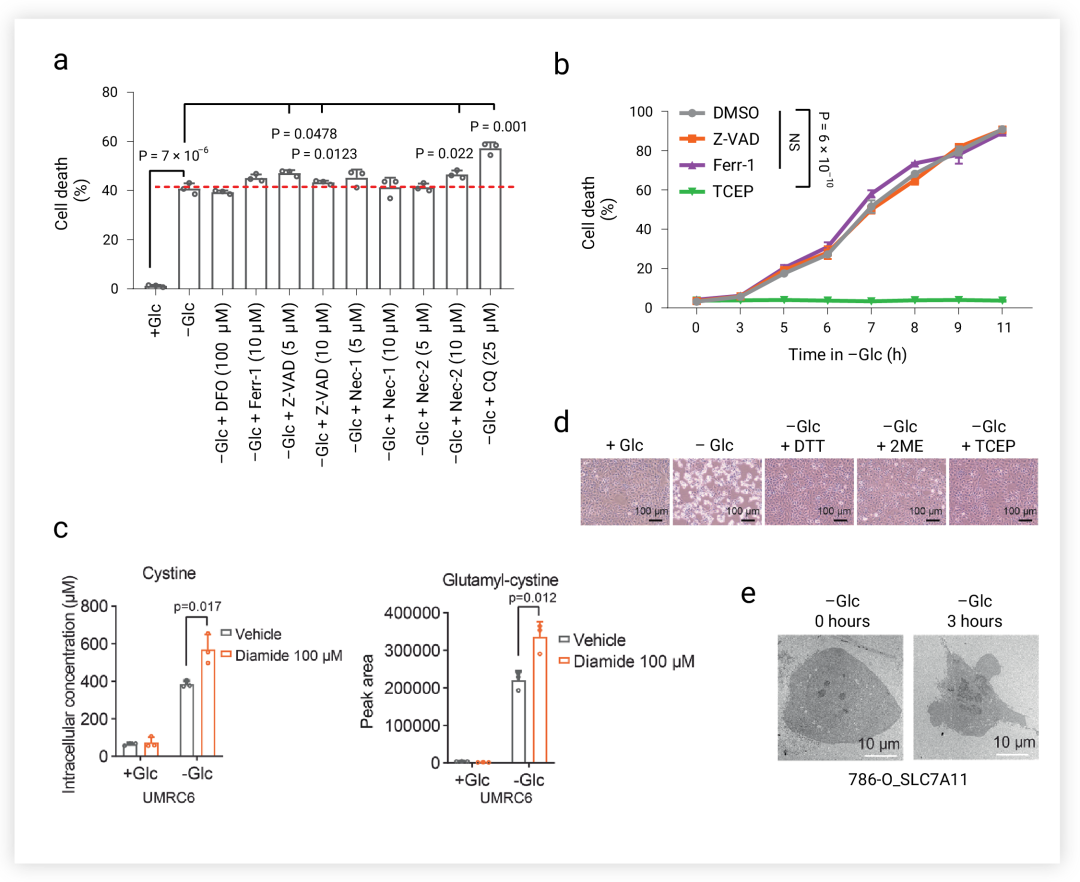
双硫死亡是一种不同于铁死亡,凋亡等的新型死亡方式,在 SLC7A11high 细胞中,铁死亡,凋亡,细胞坏死以及自噬抑制剂都不能挽救葡萄糖饥饿诱导的细胞死亡 (图 3a-b), 但二硫应激的还原剂,如二硫苏糖醇 (DTT)、β-巯基乙醇 (2ME) 和 TCEP 可以完全抑制 SLC7A11high 细胞中葡萄糖饥饿诱导的细胞死亡 (图 3d)。此外,硫醇氧化剂(二胺和马来酸二乙酯)促进 SLC7A11high 细胞在葡萄糖饥饿下的细胞死亡,并导致细胞内二硫分子的急剧积累(如胱氨酸和谷氨酰胱氨酸,经二胺处理后进一步增加) (图 3c)。透射电镜分析显示,在葡萄糖饥饿导致 SLC7A11high 细胞种胱氨酸在细胞质中积聚 (图 3e)。以上结果表明二硫胁迫引起的细胞死亡,与铁死亡,细胞凋亡等都不同,那么其特征到底是什么?
图 3. 葡萄糖饥饿条件下的细胞死亡方式[5] a. SLC7A11-high 细胞的用 DFO, Fer-1, Z-VAD, Nec-1, Nec-2 和 CQ 处理后的细胞死亡情况。b. 过表达 SLC7A11 的细胞在无葡萄糖培养基中培养中用 Z-VAD, Fer-1 和 TCEP 处理指定时间。c. UMRC6 细胞在含或不含 DTT, 2ME 或 TCEP的培养基中培养。d. UMRC6 细胞内胱氨酸和谷氨酰胱氨酸的积累。e. UMRC6 细胞的典型透射电镜图像。 双硫死亡---与肌动蛋白细胞骨架有关
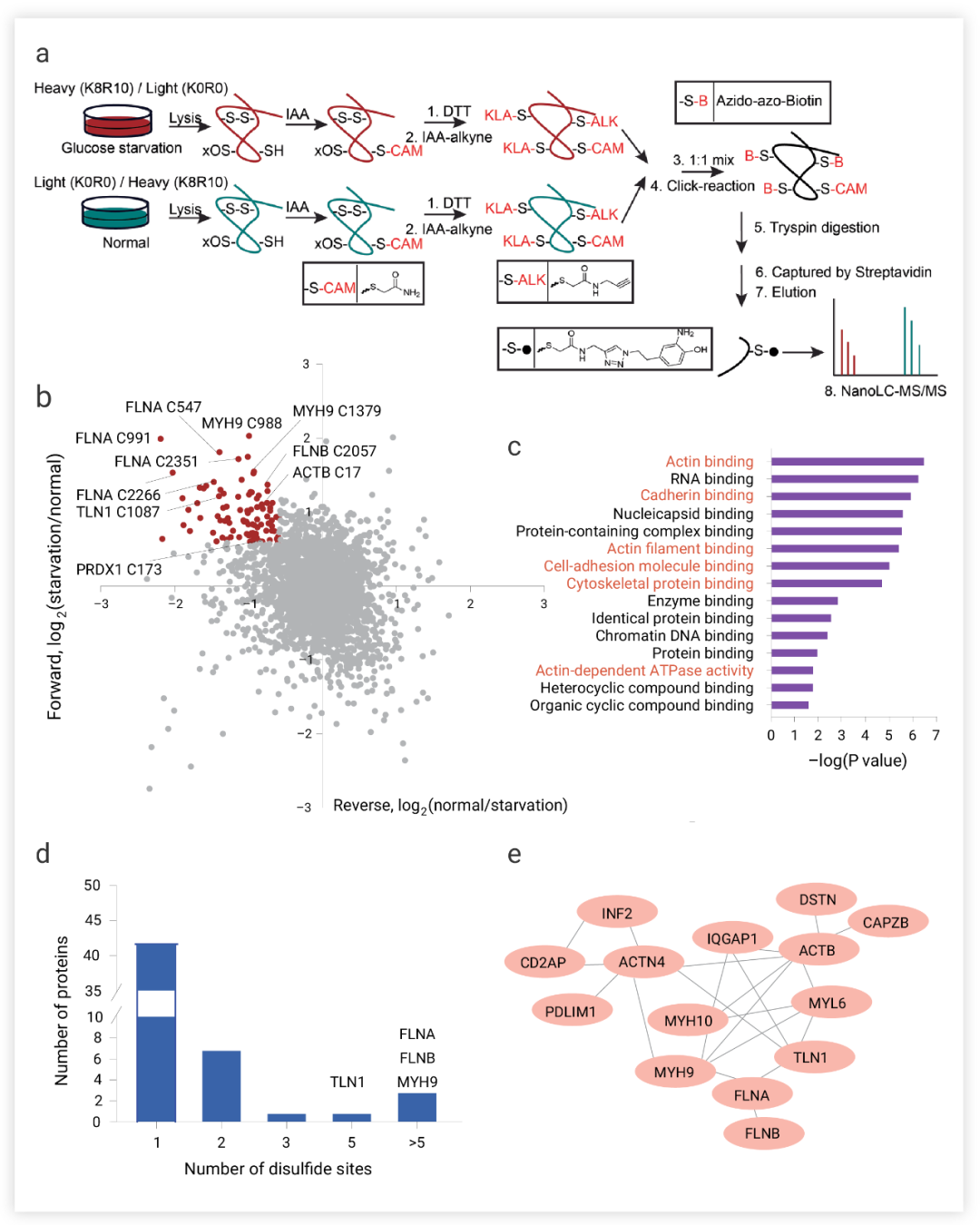
作者团队假设,在葡萄糖饥饿条件下,SLC7A11 高细胞的 NADPH 消耗和二硫应激的增加诱导氧化还原敏感蛋白中二硫键的生成 (在正常条件下,细胞质的还原环境阻止胞质蛋白形成二硫键),这可能会破坏相应的氧化蛋白的活性或功能,从而损害细胞活力。为了验证这一假设,作者团队对氨基酸进行稳定同位素标记,量化葡萄糖饥饿诱导的 SLC7A11high 细胞中的二硫化物蛋白质组改变 (图 4a)。正向和反向标记分析确定了 90 个半胱氨酸位点。此外,基因本体分析表明,在葡萄糖饥饿诱导的二硫键的蛋白质中,肌动蛋白细胞骨架和细胞粘附相关的生物过程或途径显著富集 (图 4c),作者团队还发现了至少 17 个肌动蛋白细胞骨架蛋白在葡萄糖饥饿后二硫键增加的蛋白中 (图 4b) 并且这些蛋白质中的大多数包含二硫键的半胱氨酸位点 (图 4d-e)。这表明 SLC7A11high 细胞中的葡萄糖饥饿可能诱导肌动蛋白细胞骨架蛋白中的二硫键 (图 4c)。 图 4. 葡萄糖饥饿条件下肌动蛋白细胞骨架蛋白中的二硫键形成[5] a. 用于鉴定含二硫肽的方法 正向和反向实验中含二硫肽的散点图。c. 基因本体 (GO) 富集分析。d. 含有二硫键的蛋白质的不同含二硫半胱氨酸位点的数量的增加。
SLC7A11-high 细胞的肌动蛋白细胞骨架动力学
由于二硫键的形成会影响非还原条件下蛋白质的电泳迁移率。作者团队检测了肌动蛋白细胞骨架蛋白的迁移率,发现 UMRC6 细胞在葡萄糖饥饿后,多个肌动蛋白细胞骨架蛋白表现出较慢的迁移,这表明这些肌动蛋白细胞骨架蛋白在葡萄糖饥饿条件下形成多个分子间二硫键 (图 5a) 。作者团队还进一步研究葡萄糖饥饿后 SLC7A11high 细胞的肌动蛋白细胞骨架动力学。在含糖培养基的 SLC7A11high 细胞中,肌动蛋白丝 (F-肌动蛋白) 主要在细胞皮质和应激纤维中;但葡萄糖饥饿会诱导细胞形态的显著变化:细胞收缩和 F-肌动蛋白收缩。F-actin 与膜染料 CellMask 共染色显示,葡萄糖饥饿会导致 SLC7A11high 细胞中 F-肌动蛋白从质膜分离 (图 5b-d),并且葡萄糖饥饿诱导的这些细胞肌动蛋白细胞骨架形态的变化是 SLC7A11 依赖性的 (图 5c),胱氨酸饥饿 、2DG 或 2ME (图 5f) 处理可以消除这种变化。葡萄糖饥饿诱导的 SLC7A11 高细胞肌动蛋白骨架蛋白异常二硫键可能导致随后的 F-肌动蛋白收缩和脱离质膜。
图 5. 葡萄糖饥饿条件下,异常二硫键形成导致的肌动蛋白动力学[5] a-b. 谷胱甘肽化 slc7a11-high 介导的胱氨酸摄取示意图。c. WT 和 SLC7A11-KO UMRC6 细胞的无糖培养基中对 F-肌动蛋白进行荧光染色。d. 无糖培养基中培养的细胞的 F-肌动蛋白和膜进行荧光染色。e. 含葡萄糖、无葡萄糖、葡萄糖和无胱氨酸 (−Glc) 或无胱氨酸 (−Glc) 培养基中培养,对 F-肌动蛋白进行荧光染色。F. 2ME 添加在含糖或无糖培养基中培养的细胞中 F-肌动蛋白的荧光染色。
以上结果表明,SLC7A11high 细胞中,葡萄糖饥饿诱导肌动蛋白细胞骨架蛋白的异常二硫键,F-肌动蛋白以 SLC7A11 依赖性的方式崩溃。新的死亡机制的鉴定促进人们对细胞稳态的基本理解,那 “双硫死亡” 最大的意义何在?
双硫死亡,研究的意义何在?
葡萄糖是糖酵解的起始物质,由葡萄糖转运蛋白 (GLUT) 家族通过细胞膜转运,靶向葡萄糖转运蛋白 (GLUT) 是是潜在癌症治疗干预的有趣靶点。甘波谊团队等人在 2020 年的发表的文章中就发现高 SLC7A11 表达的癌细胞对葡萄糖转运蛋白 GLUT 抑制剂特别敏感。GLUT 抑制剂 KL-11743 或 Bay-876 可有效抑制葡萄糖摄取,与葡萄糖饥饿情况相似。在 SLC7A11 过表达细胞中,GLUT1 抑制剂处理增加 NADP+/NADPH比值 (图 6a,b),并在 UMRC6 细胞中引发强烈的细胞死亡(图 6c)。此外,GLUT 抑制会诱导二硫键的结合肌动蛋白骨架蛋白和 F-肌动蛋白网络崩溃。在动物模型中,Bay876 治疗降低了 SLC7A11high NCI-H226异种移植瘤的生长 (图 6d),Bay -876 处理的肿瘤表现出频繁的细胞死亡 (图 6e),Bay-876 处理的肿瘤在肌动蛋白细胞骨架蛋白中表现出更多的二硫键结合。这些结果表明:GLUT 抑制剂诱导 SLC7A11high 癌细胞的双硫状态和细胞死亡,而癌细胞的双硫死亡可能是介导 GLUT 抑制剂治疗 SLC7A11high 肿瘤的治疗效果的关键因素。
图 6. GLUT 抑制剂诱导 SLC7A11 高表达细胞死亡[5]a,葡萄糖摄取水平。b.NADP+/NADPH比值 c. 死亡细胞的比例。BAY-876 与DFO, fer-1,Z-VAD、 Nec-1、Nec-2 和 CQ 在指定浓度下处理 7h。d. NCI-H226 异种移植模型中肿瘤体积随时间的变化。e. NCI-H226 异种移植物的重量以及 NCI-H226 肿瘤区域的 HE 染色和免疫组化染色。
■ 小结
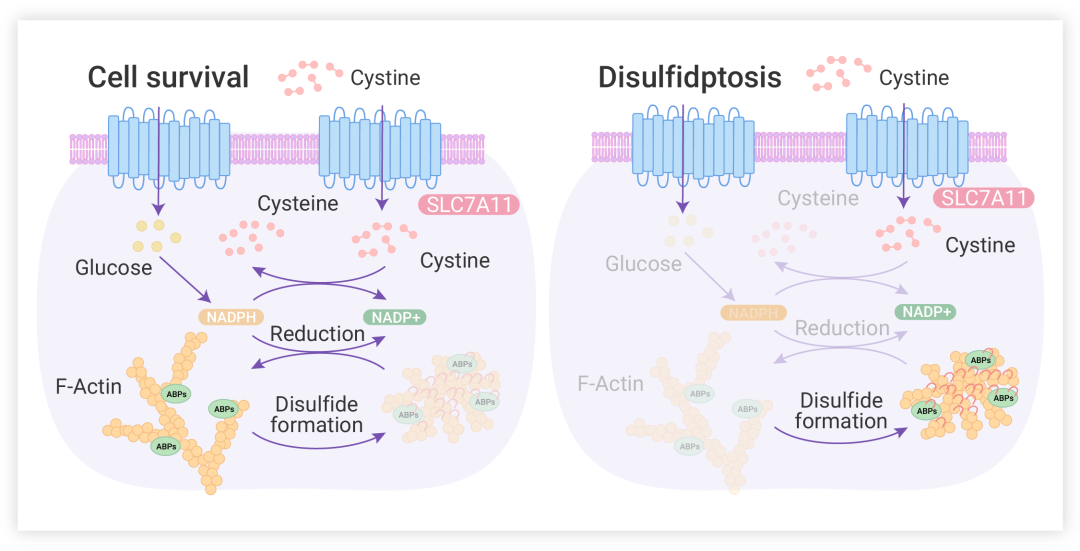
作者团队研究结果表明在 SLC7A11 高表达的情况下,葡萄糖饥饿限制 PPP 产生 NADPH 会导致小分子二硫化物 (包括胱氨酸) 大量积累、引起一系列氧化还原缺陷和细胞死亡。
图 7. 双硫死亡的机制图
并且基于对双硫死亡的机制理解,还发现 GLUT 抑制诱导的双硫死亡可能是治疗slc7a11 高表达肿瘤的有效治疗策略,这种肿瘤经常发生在人类癌症中[6]。双硫死亡这种独特的细胞死亡机制的阐明为靶向治疗癌症提供一个关键框架。
MCE 的所有产品仅用作科学研究或药证申报,我们不为任何个人用途提供产品和服务 参考文献 1. Wenxin Song, et al. Solute carrier transporters: the metabolic gatekeepers of immune cells. Acta Pharm Sin B. 2020 Jan;10(1):61-78. 2. Xiaoguang Liu, Yilei Zhang , Li Zhuang, et al. NADPH debt drives redox bankruptcy: SLC7A11/xCT-mediated cystine uptake as a double-edged sword in cellular redox regulation . Genes Dis. 2020 Nov 25;8(6):731-745. 3. Takeo Goji, et al. Cystine uptake through the cystine/glutamate antiporter xCT triggers glioblastoma cell death under glucose deprivation. J Biol Chem. 2017 Dec 1;292(48):19721-19732. 4. Xiaoguang Liu, Kellen Olszewski, Yilei Zhang, et al. Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat Cell Biol. 2020 Apr;22(4):476-486. 5. Xiaoguang Liu, Litong Nie, et al. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat Cell Biol. 2023 Feb 6. 6. Pranavi Koppula, Li Zhuang, Boyi Gan. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. 2021 Aug;12(8):599-620.
| 




 楼主
楼主