登陆查看更多优秀资源帖,与同道便捷交流讨论
您需要 登录 才可以下载或查看,没有帐号?立即注册
x
一般而言,一种药物研发周期在 10 年以上,研发投入在数十亿美金,并且呈现逐年上升的趋势。近年来,深度学习 (Deep Learning, DL) 技术在语音识别、图像识别等领域取得重大突破,迅速成为学术界和工业界的研究热点。
深度学习是人工智能的一个分支,它是一种利用神经网络进行学习的技术。深度学习技术在生物医学领域已经取得重要进展,目前研究人员已经开发了一系列基于深度学习的疾病诊断、蛋白质设计、医学图像识别的应用策略。制药工业界目前也开始重视深度学习技术,希望利用其加速药物研发并降低成本。

此前的研究表明,深度学习技术在优化化学合成路线、预测药物的药代动力学性质、预测药物的作用靶点以及生成新型分子等方面具有优势。
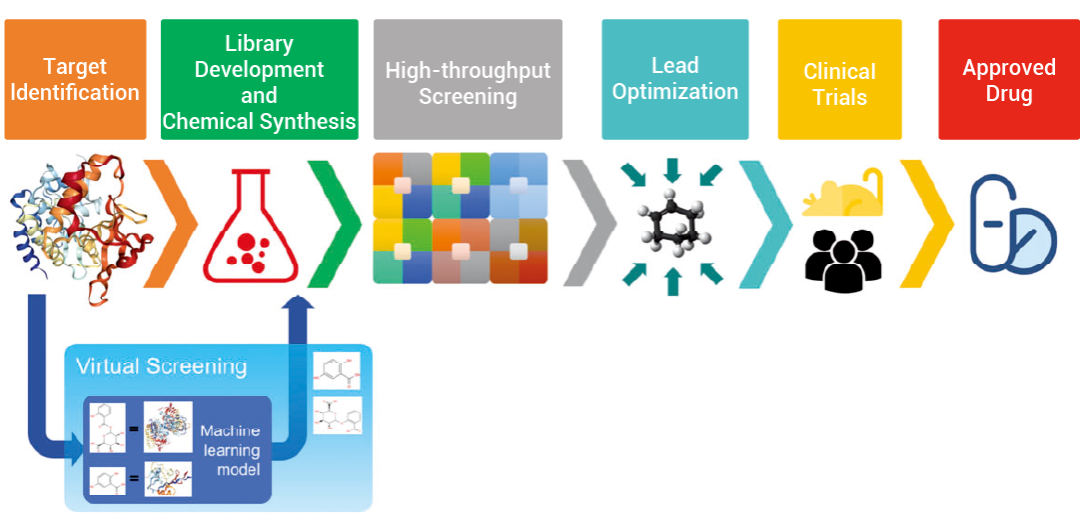
图 1. 虚拟筛选在药物开发流程中的地位[1]。 ▐ 虚拟筛选:化合物-靶蛋白的亲和力
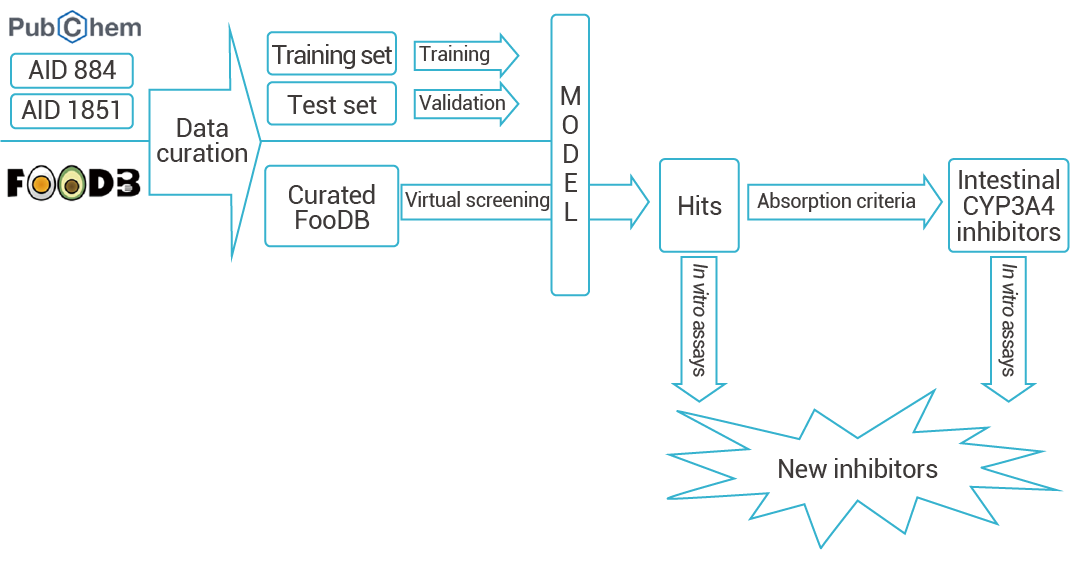
深度学习可以通过训练大量的已知化合物-靶蛋白相互作用数据来学习化合物和靶蛋白之间的内在关系。这种训练过程使得深度学习模型能够自动提取和利用化合物和靶蛋白的特征,以及它们之间的相互作用模式。Yelena Guttman 等人基于 DeepChem 框架,构建了一个 CYP3A4 抑制剂预测模型。先基于 lipinski 五原则对库中化合物进行排除,再基于此模型对 FOODB 库中 68,900 个化合物进行 CYP3A4 抑制活性预测,顺利得到了两种新的 CYP3A4 抑制剂。
图 2. 基于 DeepChem 的 CYP3A4 抑制剂预测[2]。 在 KNIME 分析平台 4.0.314 中创建了一个工作流来准备和分析虚拟筛选。
▐ 预测化合物的 ADMET 性质
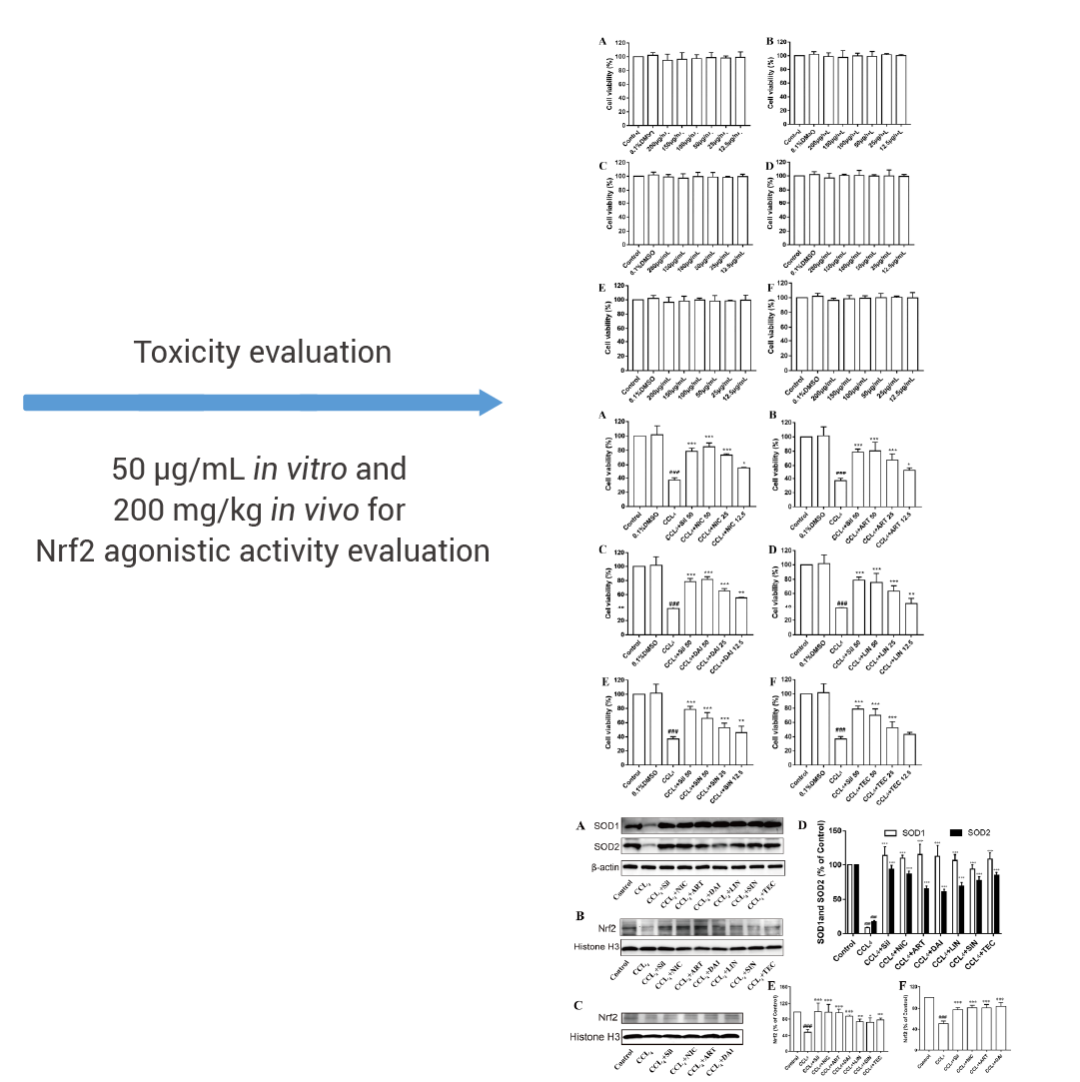
自 Lipinski 类药五原则提出以来,对先导化合物的 ADMET (吸收、分布、代谢、排泄和毒性) 性质进行早期预测也变得越来越重要。许多研究表明,通过对大量已知化合物的 ADMET 数据进行训练和学习,自动识别和提取化合物特征与性质之间的关系,训练好的深度学习模型可以用来预测新化合物的性质,从而加速药物发现和开发的进程。Liu 等人利用定向消息传递网络 (directed message passing neural networks, D-MPNN,又称 Chemprop) 对 FOODB 库中化合物进行了 Nrf2 激动活性预测及毒性分析,顺利得到了Nicotiflorin这一兼具 Nrf2 激动活性和安全性的药物,并且在体内外实验中得到了验证[3]。
图 3. 利用深度学习算法评估药物安全性[3]。
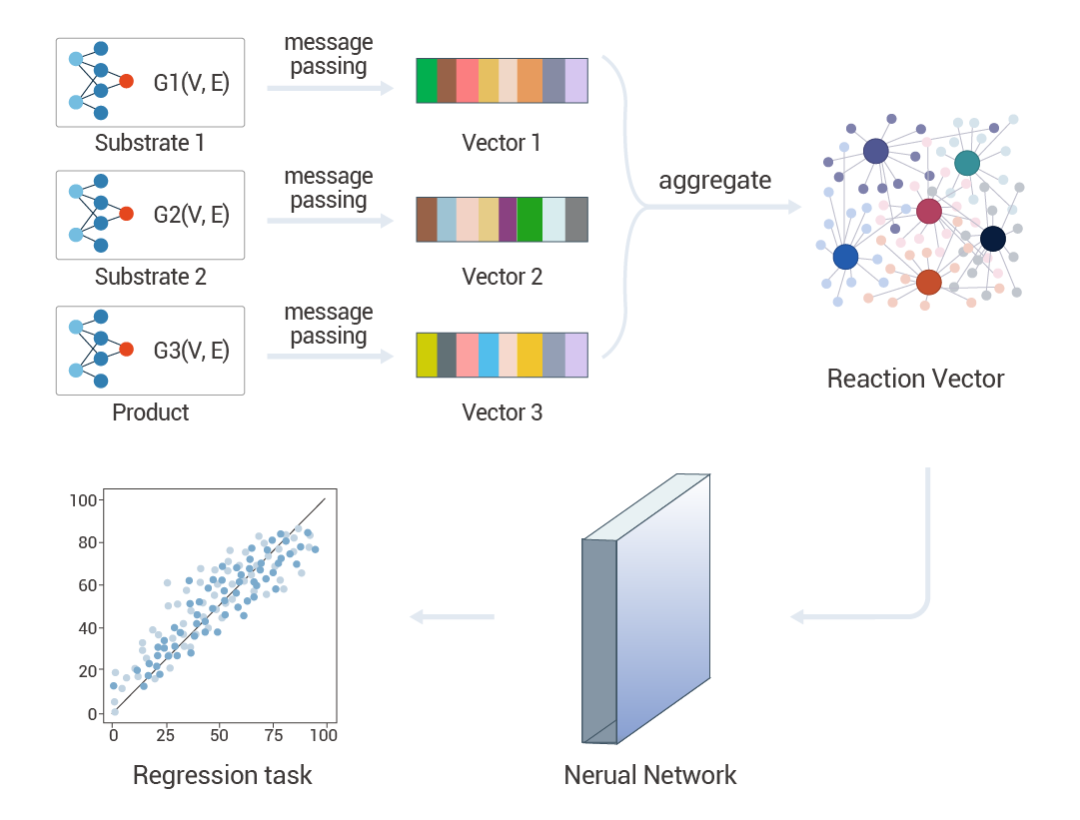
▐ 优化化学合成路线近年来,人们已经看到人工智能 (AI) 开始为化学合成带来革命性的变化。然而,由于缺乏合适的化学反应表示方式和反应数据的稀缺性,限制了人工智能在反应预测中的广泛应用。深度学习可以通过对大量的化学合成数据进行训练和学习,自动识别和提取合成路线的特征和模式,用来预测新的合成路线的效率和选择性,从而加速新药的开发和生产。
图 4. 深度学习在预测化学反应合成路线上的应用[4]。 Schwaller 等人结合了深度学习网络和符号人工智能来规划化学合成路线。他们开发了一个名为“MoleculeNet”的框架,该框架能够预测反应是否可能成功,并使用这些预测来规划出从起始原料到目标分子的合成路径[5]。

深度学习在虚拟筛选领域中的应用,主要是通过神经网络来预测化合物的活性或性质,从而在虚拟环境中筛选出有潜力的候选药物或材料。
以下是一些常见的深度学习算法在虚拟筛选中的应用:
卷积神经网络 (CNN):CNN 特别适合处理图像数据,如分子结构图。通过识别和提取分子中的特征,如原子和化学键的类型和位置,CNN 可以预测分子的性质和活性。
循环神经网络 (RNN):对于处理序列数据 (如化学分子序列) 的虚拟筛选任务,RNN 特别有用。RNN 可以捕捉分子序列中的长期依赖关系,从而更准确地预测分子的性质。
生成对抗网络 (GAN):GAN 可以生成新的分子结构,这在进行虚拟筛选时非常有用。通过训练 GAN,可以生成具有所需性质的分子,从而大大减少实验的必要性。
图神经网络 (GNN):GNN 特别适合处理图结构数据,如分子图。GNN 可以捕捉分子中原子和化学键之间的关系,从而更准确地预测分子的性质。
Transformer:对于处理长序列数据的虚拟筛选任务,如多步化学反应预测,Transformer 是一个很好的选择。Transformer 可以捕捉序列中的长期依赖关系,从而更准确地预测分子的性质。
今天,小 M 给大家介绍了深度学习在药物研发领域的应用方向及常见算法,作为一种新兴的技术,AI / 深度学习技术在新药研发领域已初见成效,相信随着科学的进步,AI 助力药物筛选一定会在生物医药领域有着更加深远的影响。

Virtual Screening
虚拟筛选 (Virtual Screening, VS) 是基于小分子数据库开展的活性化合物筛选。利用小分子化合物与药物靶标间的分子对接运算,虚拟筛选可快速从几十至上百万分子中,遴选出具有成药性的活性化合物,大大降低实验筛选化合物数量,缩短研究周期,降低药物研发的成本。
| MCE 50K Diversity Library
MCE 50K Diversity Library 由 50,000 种类药化合物组成。依据谷本相似性 (Tanimoto Coefficient) 及聚类算法 (Bemis-Murcko) 对上百万化合物进行筛选以确保结构多样性。本多样性库具备新颖性、类药性,化合物结构类型多样、化学空间丰富,库中化合物可重复供应,是新药研发的有力工具,可以广泛地应用于高通量筛选 (HTS) 和高内涵筛选 (HCS)。
| MegaUni 10M Virtual Diversity Library
运用生成式人工智能技术,依托强大的计算能力,基于高质量的 40,662 个分子砌块,匹配合适的反应规则,选择最优的化合物生成策略,去除合成难度高、类药性低、PAINS 等不利化合物后,进一步分析化合物骨架,优选出类药多样性分子组成虚拟库,适用于 AI 药物筛选、大型虚拟筛选等。
| MegaUni 50K Virtual Diversity Library
Retatrutide 是胰高血糖素受体 (GCGR)、葡萄糖依赖性促胰岛素多肽受体 (GIP 优选 50,000 个分子组成 MegaUni 50K Virtual Diversity Library。50,000 个分子具有 46,744 种 BMS 分子骨架,每种分子骨架仅包含 1-3 个化合物,化学空间多样,结构新颖,适用于新型先导物发现等。
|
MCE 的所有产品仅用作科学研究或药证申报,我们不为任何个人用途提供产品和服务。
参考文献:
[1] Rifaioglu AS, et al. Recent applications of deep learning and machine intelligence on in silico drug discovery: methods, tools and databases. Brief Bioinform. 2019 Sep ;20(5):1878-1912.[2] Guttman Y, Kerem Z. Dietary Inhibitors of CYP3A4 Are Revealed Using Virtual Screening by Using a New Deep-Learning Classifier. J Agric Food Chem. 2022 Mar ;70(8):2752-2761.
[3] Liu S, et al. Virtual Screening of Nrf2 Dietary-Derived Agonists and Safety by a New Deep-Learning Model and Verified In Vitro and In Vivo. J Agric Food Chem. 2023 May ;71(21):8038-8049.
[4] Li B, et al. A deep learning framework for accurate reaction prediction and its application on high-throughput experimentation data. J Cheminform. 2023 Aug;15(1):72.
[5] Segler MHS, et al. Planning chemical syntheses with deep neural networks and symbolic AI. Nature. 2018 Mar ;555(7698):604-610.
| 




 楼主
楼主