登陆查看更多优秀资源帖,与同道便捷交流讨论
您需要 登录 才可以下载或查看,没有帐号?立即注册
x
作者:南方医科大学南方医院血液科 周红升、孟凡义、黄芬等;病理科 邓永键
性别:男
年龄:45
病史:因间断发热、咳嗽气促伴肺部阴影1年余于2006年12月28日入住南方医科大学南方医院血液科。
临床资料:患者2005年1月体检胸部X线检查发现右肺阴影,腹部CT正常,血常规示白细胞7.83×109L,嗜酸粒细胞(EOS)0.81×109/L,因无不适未诊治。2月初出现咳嗽,咯黄痰,自服感冒药(具体不详)后好转。下半年起间断午后发热,体温最高38.5℃,伴畏寒,咳嗽咯痰无加重,无其他不适,自服对乙酰氨基酚(百服咛)可以缓解。年底出现气促,活动后明显,体重减轻(具体不详),未诊治。2006年5月初无明显诱因突发高热,体温39.2℃,气促加重,血常规示白细胞正常,EOS比例增高,X线胸片示双肺炎症,B超示脾大,予青霉素静脉滴注4d后体温恢复正常,但咳嗽咯痰、肺部病灶无改变,当再次发热时,纤维支气管镜(纤支镜)检见右中叶支气管狭窄,镜下肺活检病理学示淋巴细胞浸润性炎症,予抗感染治疗(具体不详)无效。6月初查血常规EOS1.5×109/L,球蛋白49g/L,IgM23.8g/L,肝吸虫、弓形虫阳性,再次经纤支镜肺活检结果大致同前,先后予头孢他啶、阿奇霉素、病毒唑、氟康唑、莫西沙星、复方磺胺甲恶唑片足疗程抗感染治疗后仍低热,肺部阴影先有吸收后又进展。加用糖皮质激素治疗(甲泼尼龙40mg/d×2→泼尼松30mg/d,后逐渐减量),体温持续正常,气促逐渐消失,EOS降至正常,肺部病灶明显吸收,诊断“肺嗜酸粒细胞浸润症(PIE)”。2006年7月骨髓涂片示分类不明细胞26%,当泼尼松减量到10mg,患者病情反复。2006年10月外周血EOS增高,球蛋白38g/L,IgM16.98g/L,抗核抗体(ANA)阳性,余自身免疫抗体、抗中性粒细胞胞质抗体(ANCA)、抗心磷脂抗体、抗肾小球基底膜抗体阴性;纤支镜检查提示右中叶支气管、右下前基底支支气管铺路石样改变,支气管肺泡灌洗(BAL)和纤支镜刷片均可见大量淋巴细胞。经纤支镜肺活检可见大量淋巴细胞(图1),少量浆细胞、组织细胞浸润,未见明确EOS。多科室会诊后考虑结缔组织病可能性大,继续给予激素治疗(甲泼尼龙0.5g/d×3→口服泼尼松50mg/d减量至30mg/d维持),气促缓解,肺部病灶缩小,EOS降至正常。同年12月中旬反复出现肌肉酸痛,门诊査球蛋白72.1g/L,血清免疫固定电泳示IgM-Kappa型条带显著增高,骨髓细胞涂片示原始幼稚浆细胞占26%,外周血原始幼稚浆细胞占30%,门诊以“巨球蛋白血症浆细胞白血病?”收人血液科。自起病以来,精神及睡眠差,体重下降约4kg。既往体健。
[align=center][align=center][align=center] 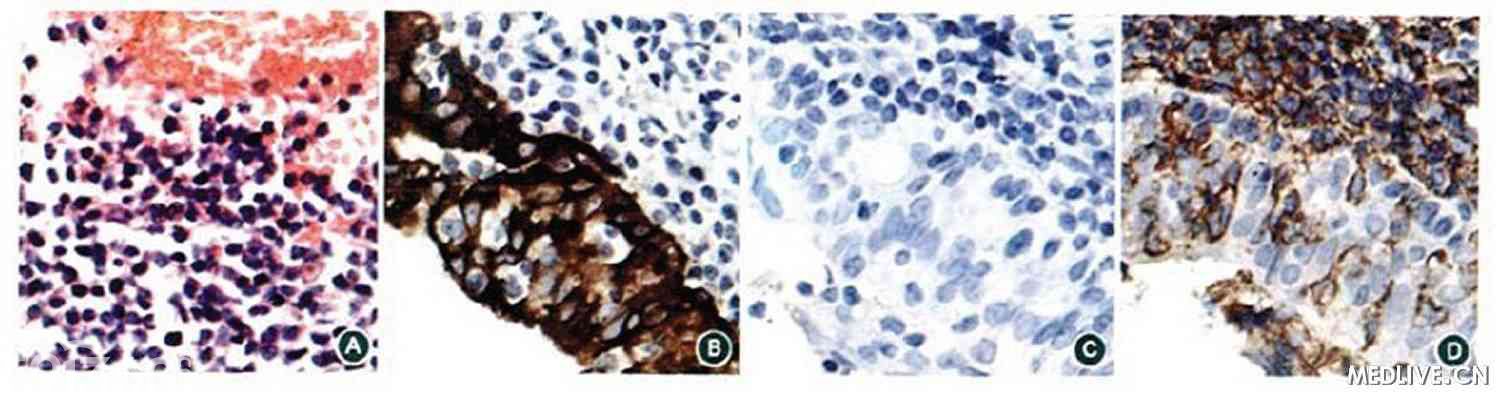 [/align][/align][/align]
A.肺组织见大量小淋巴细胞浸润HE;
B.肺组织CK染色阳性;
C.小淋巴细胞CyclinD1阴性;
D.小淋巴细胞弥漫表达CD20。
查体:生命体征正常,轻度贫血貌,全身浅表淋巴结未触及,胸骨无压痛,右中下肺可闻及少许velcro音,脾肋下6cm,质地硬,无触痛,肝脏肋下未们及,其余无异常。
辅助检査:
外周血:白细胞18.4×109/L,原幼浆细胞30%,血红蛋白97g/L,血小板253×109/L;血清球蛋白72.1g/L,IgM60.7g/L,β2-MG6.63mg/L;抗ANA阳性;血清免疫固定电泳:IgM-Kappa单克隆球蛋白异常增高。
骨髄细胞涂片:增生活跃,原幼浆细胞占11.0%,分类不明细胞占22.0%(呈散在或灶状分布)(图2)。
[align=center][align=center][align=center] 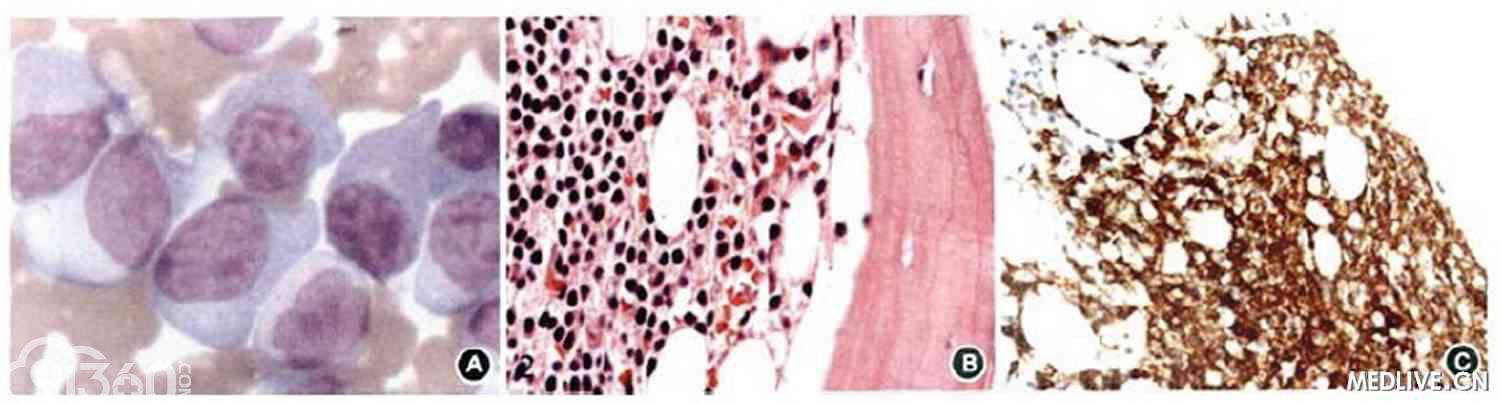 [/align][/align][/align]
A.骨髓细胞瑞氏染色,可见胞体大、胞质丰富、染色质粗密、单核或双核的浆细胞×1000;
B.单核样小淋巴细胞HE染色,呈新月状浸润骨髓骨小梁组织×400;
C.浸润小淋巴细胞免疫组织化学染色,可见CD20弥漫阳性×400;
D.浸润小淋巴细胞免疫组织化学染色,可见CD79a弥漫阳性×400。
骨髓活检病理:网硬蛋白( );免疫组化:CD38(-),CD138(-),CD20(淋巴样细胞弥漫 ),CD79a(淋巴样细胞弥漫 ),CD45RO(淋巴样细胞散在 ),CD3(淋巴样细胞散在 ),CD34(-),CD61(巨核细胞 ),见片状分布的幼稚B小淋巴细胞浸润(图2)。
流式细胞术分析:病态细胞R1群占28.8%,CD45弱阳性,表达CD19,CD138,CD38,HLA-DR,R2群病态细胞占37.8%,CD45阳性,表达CD19,CD20,CD22,CD25,HLA-DR,CD380。
B超示:肝脏增大;肝内胆管结石,脾脏增大:腹膜后未见明显肿大淋巴结。胸、腰椎、骨盆、颅骨骨质X线检查未见异常。
CT检查:胸部CT示双肺弥漫性渗出性病变(图3)。
[align=center][align=center][align=center] 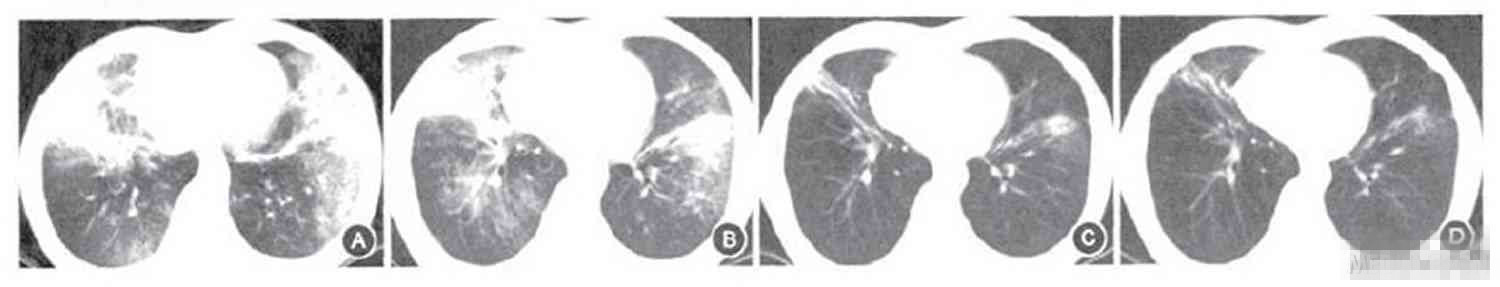 [/align][/align][/align]
A.2006年10月13日肺部CT,可见双肺弥漫渗出性病变;
B.2006年11月8日肺部CT,可见双肺病灶较前有所吸收;
C.2007年9月4日肺部CT,可见双肺病灶明显吸收,尚残留条索状阴影;
D.2008年10月21日肺部CT,可见条索状机化病灶较前无明显改变。
诊断过程
患者入院诊断首先考虑“巨球蛋白血症,浆细胞白血病?”,但患者病情进展惰性,无骨痛、骨质破坏等骨髓瘤病程,不支持浆细胞白血病诊断;骨髓细胞学及流式免疫分型提示为反应性浆细胞,结合临床特征及辅助检查,考虑为反应性浆细胞增加,继发性巨球蛋白血症,需鉴别“淋巴瘤、慢性感染/寄生虫感染、PIE、结缔组织病”。
经多科室联合会诊,意见如下:
呼吸科认为:患者由右肺孤立性病灶伴EOS增高发展为弥漫性肺间质病变,多次BAL、肺活检排除了PIE,需要排除结缔组织病及淋巴瘤样肉芽肿(LYG);
风湿科意见:患者仅ANA阳性,伴弥漫性肺损害,考虑结缔组织病如多发性肌炎/皮肌炎(PM/DM)肺累及,证据不足;
病理方面:两次肺活检均见较成熟小淋巴细胞弥漫浸润,胞质丰富,呈单核样,伴少量浆细胞、组织细胞浸润,免疫组化示CD20 ,CD5-CD10-CD23-CyclinD1-;
骨髓活检:见局灶性单核样小淋巴细胞浸润骨小梁,偶见浆细胞,免疫组化示CD20 CD79a CD138-,少数呈浆样分化(CD38 ),考虑为淋巴瘤浸润骨髓;
病理类型上:活检组织内未见具有胞体小、核偏位、伴Dutcher小体及肥大细胞浸润等特征的浆细胞样淋巴细胞,不支持华氏巨球蛋白血症/淋巴浆细胞样淋巴瘤(WM/LPL),最后病理诊断为肺黏膜相关淋巴组织结外边缘区淋巴瘤(MALT淋巴瘤),白血病期。
血液科总结认为:患者骨髓细胞学检査可见分类不明细胞及典型的浆细胞,流式免疫分型分析两群细胞分别成熟B淋巴细胞和浆细胞来源,结合形态学和免疫学分析,不支持WM/LPL诊断;临床特征上,以孤立性右肺阴影起病,逐渐发展为双肺弥漫性改变,后期(1年半后)方出现球蛋白增加及单克隆IgM,骨髓见浆细胞比例增加,临床表现符合伴浆样化生的肺MALT淋巴瘤进展期侵犯骨髓、继发球蛋白及浆细胞增加,病情演变上不符合WM/LPL的特征。
治疗:
确定诊断后分别予CHOP(环磷酰胺 阿霉素 长春新碱 泼尼松)、氟达拉滨-CHOP方案各化疗2个疗程后,患者体温正常,咳嗽气促消失,骨髓浆细胞降至9%,幼稚淋巴细胞9.5%;继用ACE(阿糖脆苷 顺铂 依托泊苷)方案化疗1个疗程,肝脾回缩至正常,骨髓获完全缓解;后ACE方案巩固4个疗程。
2007年8月起停化疗,PET-CT示完全缓解,随访2010年2月,复査肺部CT示右肺中叶残留条索状病灶,其他治疗前异常指标持续正常。
诊治难点:
1.肺MALT淋巴瘤临床罕见,常以孤立性肺部占位为首发症状可发展至弥漫性双肺损伤、骨髓和肝脾浸润。
2.肺MALT淋巴瘤易侵犯骨髓,病理缺乏特异性,与WM/LPL鉴别困难。
启示:
1.临床上对于抗感染无改善、激素治疗有效、结缔组织病证据不足的肺部阴影,应警惕肺淋巴瘤的可能,及时进行病理活检。
2.肺MALT淋巴瘤与WM/LPL应结合临床和病理进行鉴别,荧光原位杂交或PCR检测发现t(11;18)染色体易位产生的API2-MALT1融合基因有助鉴别。
3.肺MALT淋巴瘤预后良好,长期生存率可达70%~90%,治疗手段包括手术切除、抗CD20单抗及联合化疗,伴骨髓等结外浸润的Ⅲ~Ⅳ期患者可能需更强烈治疗。
分析与讨论:
肺淋巴瘤是临床上罕见的一类淋巴瘤,仅占肺恶性肿瘤的0.5%~1%,占非霍奇金淋巴瘤的1%,其中以MALT淋巴瘤最为常见,约占70%~90%。
临床上,肺MALT淋巴瘤多以无症状的肺部单发或多发阴影起病,男性多见,进展惰性,可逐渐出现咳嗽、气促、呼吸困难等症状。与胃肠道MALT淋巴瘤病变相对局限形成对比的是,非胃肠道MALT淋巴瘤更多以弥散起病,淋巴结、骨髓浸润比例分别可达20%和25%,而其中肺MALT淋巴瘤骨髓浸润更多见。
病理形态上,典型的MALT淋巴瘤表现为MALT组织边缘区弥漫的小至中等的、胞质丰富、淡染的中心细胞样/单核样淋巴细胞浸润,伴反应性淋巴滤泡形成,周围可见反应性浆细胞、EOS浸润,部分可见典型的淋巴瘤细胞浸润、破坏上皮结构形成的淋巴上皮病变(LEL),典型免疫表型为CD20 ,IgD-,IgM ,CD5-,CD10-,Bcl6-,CyclinD1-;MALT淋巴瘤常伴有淋巴细胞质样分化,在背景细胞复杂情况下,往往与WM/LPL的浆细胞样淋巴细胞鉴别困难。
病因方面,肺MALT淋巴瘤与幽门螺旋杆菌HP感染无关,可能与自身免疫如干燥综合征有一定联系,约40%肺MALT淋巴瘤可以检测到t(11;18)(q21;q21),染色体异位形成API2-MALT1融合蛋白,具有促增殖、抗凋亡作用。
由于肺MALT淋巴瘤临床症状及病理特征缺乏特异性,早期诊断困难,需要与各类感染、结缔组织病导致的间质性肺疾病/弥漫性肺实质疾病(ILD/DPLD)、浆细胞瘤、浆细胞白血病和其他小B淋巴细胞瘤等相鉴别,结合各自疾病的临床特征、病理活检和免疫表型不难鉴别。
由于肺MALT淋巴瘤常有浆样分化和单克隆IgM,骨髓侵犯发生率髙,与WM/LPL在细胞形态、免疫表型特征部分重叠,使部分肺MALT淋巴瘤与以骨髓浸润和单克隆IgM为特征的WM/LPL鉴别困难,容易被误诊为WM/LPL,需要结合临床、病理、遗传学等因素鉴别诊断病理上,MALT淋巴瘤发生在黏膜相关淋巴组织的边缘区(MZL),肺是MALT淋巴瘤的好发部位之一,而WM/LPL多累及骨髓和淋巴结,也可结外累及,但以结外表现为原发、后浸润骨髓和肝脾者罕见;伴浆样分化的MALT淋巴瘤与WM/LPL鉴别尤为困难,WM/LPL的背景细胞特征如小淋巴细胞间可见胞体小、核偏位、部分可见Dutcher小体、周围肥大细胞浸润的浆细胞样淋巴细胞等,可协助鉴别。
骨髓侵犯方面,MALT淋巴瘤以结节浸润为主,而LPL以小梁间弥漫浸润为特征。临床特征上,肺MALT淋巴瘤早期多表现为无症状肺部占位,骨髓侵犯、球蛋白增高及单克隆IgM多发生在病程后期,不伴有髙黏滞血症;WM/LPL多发生于老年患者,常以发现球蛋白增高、单克隆IgM及骨髓侵犯起病,常伴有贫血、高黏滞血症、神经系统损害等多系统损害表现,外周血累及少见。国外回顾性分析也显示,既往报道的一些皮肤、肺等结外累及的WM/LPL病例,大部分属于MALT淋巴瘤。
目前肺MALT淋巴瘤治疗尚没有统一的规范,对于局限性病变,多参照其他惰性淋巴瘤的治疗方案,采用利妥昔单抗单药治疗、手术切除或联合化疗,对于孤立性局限病灶,也有学者主张观察而不予治疗;肺MALT淋巴瘤常伴侵犯骨髓,且预后较未侵犯骨髓者、胃肠道MALT淋巴瘤差,可能需要更强烈的化疗。肺MALT淋巴瘤总体预后良好,5年总生存率>80%;其中Ⅰ~Ⅱ期>90%,Ⅲ~Ⅳ期也可达70%。
肺MALT淋巴瘤是一种罕见的结外非霍奇金淋巴瘤,病程进展相对惰性,总生存率较长;但累及骨髓、肝脾等结外器官常见,临床表现和病理缺乏特异性表现,容易误诊,加强对其病理及分子机制的研究将有助于提高诊治水平。
【本站为非盈利学术交流平台,部分资料来源于网络,如涉及版权问题请及时联系管理员处理;所有文章仅供公益交流,不代表本站立场。欢迎提供素材、资料等,投稿邮箱:tougao@91360.com,一经采纳将给予稿费。】 | 





 发表于 2017-12-20 17:48:52
发表于 2017-12-20 17:48:52

