登陆查看更多优秀资源帖,与同道便捷交流讨论
您需要 登录 才可以下载或查看,没有帐号?立即注册
x
噬血细胞淋巴组织细胞增多症(Hemophagocytic lymphohistiocytosis,HLH),也称为噬血细胞综合征(Hemophagocytic syndrome)。是一种进展快、严重威胁生命的疾病。其特征在于过度炎症反应的无效免疫,在<18岁的跨种族人群中HLH的发病率大约为1/100000。HLH可能被忽视,使得其发病率和死亡率较高。一些因素包括病毒感染(29%),其他感染(20%),恶性肿瘤(27%),风湿病(7%)和免疫缺陷综合征(6%)与原发及继发HLH相关。恶性肿瘤与继发性HLH病例的比例相关(高达27%),血液系统恶性疾病是最常见的,在儿童中,HLH最常见于急性B淋巴细胞性白血病[1]。
早期认识、对于治疗方法的任何合理尝试至关重要。目前的治疗方案包括免疫抑制,免疫调节,化学治疗和生物反应修饰,其次是造血干细胞移植(骨髓移植)。最近的一些研究有助于理解HLH病理生理学,指导替代治疗方案。
噬血细胞淋巴组织细胞增多症(HLH)涉及广泛的相关疾病,包括常染色体隐性家族性HLH(FHL),家族性噬红细胞淋巴组织细胞增多症,感染相关性噬血细胞综合征和自身免疫相关性噬血细胞活化综合征(MAS)。
该疾病的最早描述之一是在1939年,Scott和Robb-Smith描述了一种通过在淋巴网状系统中增殖组织细胞吞噬红细胞的病症,并称之为“组织细胞性髓性网状细胞增多症”[2]。Farquhar和Claireaux更充分地描述了HLH,FHL的家族形式[3],两名同胞姐妹死于HLH,后来在1958年,同一家族的另一个兄弟姐妹以同样的症状出现[4]。Risdall是第一个描述与HLH病毒关联的人之一[5]。在此后的几年中,研究人员已经认识到这种疾病的广泛范围和感染常常触发原发和继发HLH的事实。不管原因如何,在生理上,HLH的特征在于细胞毒性细胞功能的恶化以及不能控制的巨噬细胞活化,导致过度的细胞因子产生、免疫失调和组织损伤。未经治疗,失调的炎症反应引起严重的中性粒细胞减少症,患者常常因细菌或真菌感染死亡。病情发病率高,死亡率高[6]。1983年的长期生存率估计低至4%。未经治疗中位生存期估计为<2个月[7]。
诊断标准:
以下八个HLH诊断标准中有五个符合:
1.发热;
2.脾肿大;
3.血细胞减少(至少两系,HGB<90g/L,PLT<100×10^9/L,中性粒细胞<1×10^9/L);
4.高甘油三酯血症和(或)低纤维蛋白原血症:空腹后甘油三酯>3mmol/L(265mg/dl),纤维蛋白原<1.5g/L。
5.铁蛋白升高>500g/L;
6.骨髓/脾/淋巴结中的噬血细胞增多;
7.低或不存在自然杀伤(NK)细胞活性;
8.升高的可溶性CD25(可溶性白介素IL-2受体))[8,9]。
发热是高IL水平的结果。脾肿大是淋巴细胞和巨噬细胞浸润的结果。可以通过高浓度的肿瘤坏死因子(TNF)-α和干扰素(IFN)-γ以及直接的血细胞吞噬来解释血细胞减少。高甘油三酯是由增加的TNF-α水平导致脂蛋白脂肪酶活性降低引起的。高铁蛋白>10,000μg/L已被证明在诊断HLH时有高的敏感性和高的特异性。
目前国际的主流学术观点认为,当患者出现持续发热、肝脾肿大和血细胞减少三联征应当怀疑HLH的可能;或者发热、全血细胞减少合并不明原因的肝衰竭应考虑HLH。也有研究者认为当患者出现不明原因发热,并伴有血细胞减少、铁蛋白显著升高时通常提示应完善与HLH诊断相关的检查[10]。
相关病例
病例1 一例成人噬血细胞淋巴组织细胞增多症引起的急性肝衰竭。
[align=center][align=center][align=center] 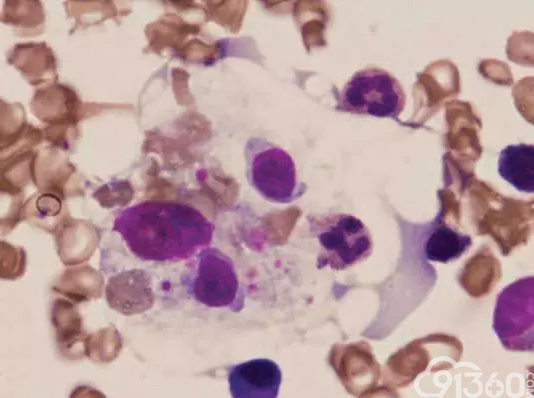 [/align][/align][/align] 图1 骨髓涂片在入院后第3天通过光学显微镜显示噬血组织细胞(Wright-Giemsa染色,×1000)
临床资料:男,34岁,恶心、疲劳持续2周,高温(最高温度高达40°C)1周,黄疸1周,严重肝损害。实验室数据显示,他患有高铁血症,血小板减少症,贫血,高甘油三酯血症和低纤维蛋白原血症。最后,骨髓活检显示噬血细胞,他被诊断患有HLH。患者用强的松和血浆置换治疗,由于病人的肝功能恶化,终于死于多器官衰竭[11]。
病例2 弥漫大B细胞淋巴瘤继发肝功能衰竭表现的HLH。
[align=center][align=center][align=center] 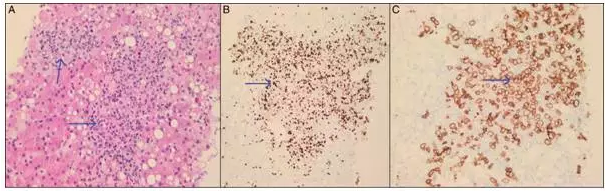 [/align][/align][/align] 图2 肝活检病理学。(A)苏木精和伊红染色(20x)显示肝脏淋巴瘤浸润。(B)Ki67(20x)显示具有高增殖指数(大于90%)的弥漫性大B细胞淋巴瘤。(C)显示弥漫性大B细胞淋巴瘤的CD20阳性表达(20x)。
临床资料:男,57岁,疲劳、眩晕、轻度黄疸。实验室检查显示贫血,血小板减少症,急性肝损伤和急性肾衰竭需要间歇性血液透析。由于乳酸性酸中毒,败血症和肝性脑病恶化,他被转移到医疗重症监护室(ICU)。血红蛋白7.5g/dL,血小板26,000/mm3,肌酐4.34mg/dL,总胆红素19mg/dL,丙氨酸氨基转移酶395U/L,天冬氨酸氨基转移酶261U/L,血清乳酸10mmol/L,血清铁蛋白>40,000ng/mL,血清甘油三酯556mg/dL。INR4.2和纤维蛋白原<70mg/dL。符合发热,双侧关节炎,脾肿大,铁蛋白水平和高甘油三酯血症的HLH标准。使用地塞米松,异环磷酰胺,卡铂和依托泊苷进行淋巴瘤和HLH的急性化疗。尽管化疗和支持治疗,但仍发展为恶化的代谢性酸中毒、低血糖、休克、硬膜下出血。病人在ICU第五天死亡[12]。
病例3 15岁白人女孩,伴有EB病毒相关噬血细胞淋巴组织细胞增多症的弥漫大B细胞淋巴瘤。
[align=center][align=center][align=center] 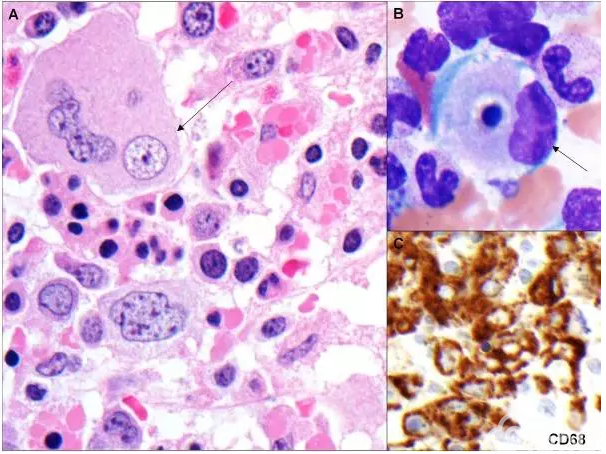 [/align][/align][/align] 图3 骨髓活检和涂片显示噬血细胞增多和CD68染色阳性的噬血细胞[13]。
病例4 疟疾相关的继发性噬血细胞淋巴组织细胞增多症。
[align=center][align=center][align=center] 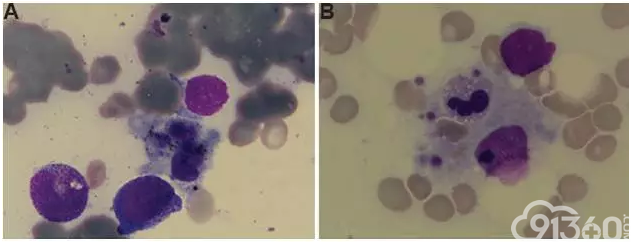 [/align][/align][/align] 图4 A骨髓抽吸涂片上观察到的噬血组织细胞B骨髓抽吸涂片,显示巨噬细胞内的白细胞,红细胞和血小板[14]。
病例5 利妥昔单抗,依托泊苷,甲基泼尼松龙,高剂量阿糖胞苷和顺铂常规化疗霍奇金淋巴瘤继发噬血细胞淋巴组织细胞增多症。
[align=center][align=center][align=center] 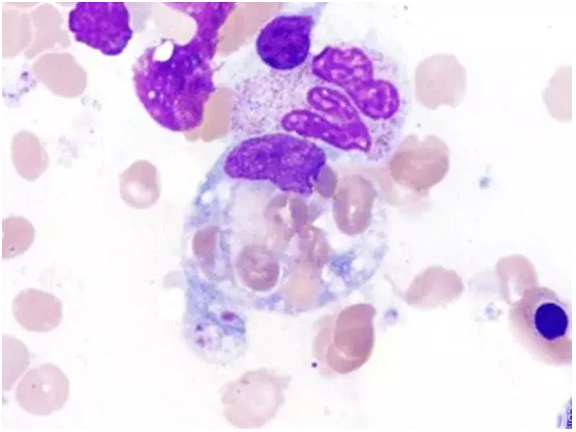 [/align][/align][/align] 图5骨髓涂片查到吞噬红细胞的噬血细胞(瑞氏-吉姆萨染色,1000×)
[align=center][align=center][align=center] 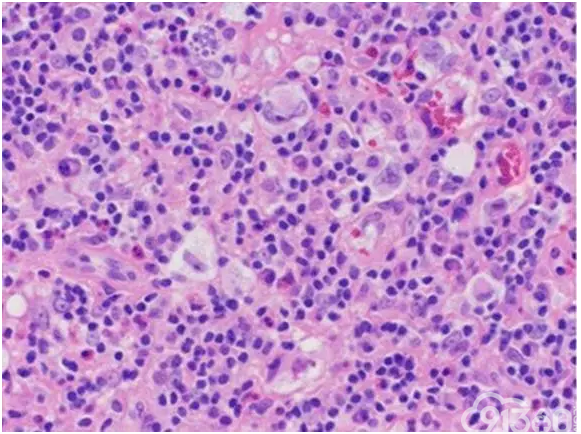 [/align][/align][/align] 图6 颈部淋巴结活检显示了混合炎症背景下的大型Reed-Sternberg细胞,诊断经典霍奇金淋巴瘤。(苏木精和曙红400×)[15]。
参考文献
[1] Melissa R George,Hemophagocytic lymphohistiocytosis: review of etiologies and management,J Blood Med. 2014; 5: 69–86.
[2] Scott RB, Robb-Smith AHT. Histiocytic medullary reticulosis. Lancet. 1939;234:194–198.
[3]Farquhar JW, Claireaux AE. Familial haemophagocytic reticulosis. Arch Dis Child.1952;27(136):519–525.
[4]Farquhar JW, Macgregor AR, Richmond J. Familial haemophagocytic reticulosis. BMJ.1958;2(5112):1561–1564.
[5] Risdall RJ, McKenna RW, Nesbit ME, et al. Virus-associated hemophagocytic syndrome: a benign histiocytic proliferation distinct from malignant histiocytosis. Cancer. 1979;44(3):993–1002.
[6] Janka G. Hemophagocytic lymphohistiocytosis: when the immune system runs amok. Klinische Padiatrie. 2009;221(5):278–285.
[7] Janka GE. Familial hemophagocytic lymphohistiocytosis. Eur J Pediatr. 1983;140(3):221–230.[PubMed]
[8] Henter JI, Horne A, Arico M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–131.
[9] Arico M, Janka G, Fischer A, et al. Hemophagocytic lymphohistiocytosis. Report of 122 children from the International Registry. FHL Study Group of the Histiocyte Society. Leukemia. 1996;10(2):197–203.
[10] 王昭,我如何诊断噬血细胞综合征,中华血液学杂志, 2016,37(07): 550-553.
[11] Acute liver failure caused by hemophagocytic lymphohistiocytosis in adults:A case report and review of the literature,Shide Lin, Ying Li, Jun Long, et al.Medicine (Baltimore)。 2016 Nov; 95(47):e5431.
[12] Patel R, Patel H, Mulvoy W,et al. Kapoor S4.Diffuse Large B-Cell Lymphoma with Secondary Hemophagocytic Lymphohistiocytosis Presenting as Acute Liver Failure.ACG Case Rep J. 2017 May 24;4:e68.
[13] Altaf S,Atreaga GM,Joshi AY,et al.Diffuse large B-cell lymphoma in an adolescent female presenting with Epstein-Barr virus-driven hemophagocytic lymphohistiocytosis: a case report.J Med Case Rep. 2012 Jun 1;6:141.
[14]Valliappan Muthu,Sahajal Dhooria, Inderpaul Singh Sehgal,et al.Malaria-associated secondary haemophagocytic lymphohistiocytosis: Report of two cases & a review of literature,Indian J Med Res. 2017 Mar; 145(3): 399–404.
[15] Hu S,Bansal P,Lynch D,et al.Rituximab, etoposide, methylprednisolone, high-dose cytarabine, and cisplatin in the treatment of secondary hemophagocytic lymphohistiocytosis with classical Hodgkin lymphoma: a case report and review of the literature.J Med Case Rep. 2016 Dec 20;10(1):365.
| 




 发表于 2017-12-20 18:12:47
发表于 2017-12-20 18:12:47

