登陆查看更多优秀资源帖,与同道便捷交流讨论
您需要 登录 才可以下载或查看,没有帐号?立即注册
x
Abstract
The clinical syndrome of primary progressive aphasia (PPA) can be associated with a variety of neuropathologic diagnoses at autopsy. Thirty percent of cases have Alzheimer disease (AD) pathology, most often in the usual distribution, which defies principles of brain-behavior organization, in that aphasia is not symptomatic of limbic disease. The present study investigated whether concomitant TDP-43 pathology could resolve the lack of clinicoanatomic concordance. In this paper, 16 cases of clinical PPA and 10 cases of primarily non-aphasic frontotemporal dementia (FTD), all with AD pathology, were investigated to determine whether their atypical clinical phenotypes reflected the presence of additional TDP-43 pathology. A comparison group consisted of 27 cases of pathologic AD with the typical amnestic clinical phenotype of probable AD. Concomitant TDP-43 pathology was discovered in only three of the FTD and PPA but in more than half of the typical amnestic clinical phenotypes. Hippocampal sclerosis (HS) was closely associated with TDP-43 pathology when all groups were combined for analysis. Therefore, the clinical phenotypes of PPA and FTD in cases with pathologic AD are only rarely associated with TDP-43 proteinopathy. Furthermore, medial temporal TDP-43 pathology is more tightly linked to HS than to clinical phenotype. These findings challenge the current notions about clinicopathologic correlation, especially about the role of multiple pathologies.
Introduction
Brain autopsies from patients presenting clinically with primary progressive aphasia (PPA), as well as behavioral and mixed variants of frontotemporal dementia (FTD), most often demonstrate frontotemporal lobar degeneration with either tau positive (FTLD-tau), TDP-43 positive (FTLD-TDP), or FUS positive (FTLD-FUS) inclusions [24, 34]. There may remain rare cases with no immunohistochemically demonstrable inclusions (dementia lacking distinctive histopathology or DLDH), but most of these have been found to have inclusions when sensitive immunostaining protocols are used [19, 22, 25]. However, as has recently been shown, many cases of PPA and FTD may have Alzheimer disease (AD) pathology at autopsy, including the typical concentration of neurofibrillary tangles within memory-related parts of the medial temporal lobes [28]. How can such a neuropathologic pattern lead to a predominantly behavioral or aphasic rather than amnestic clinical phenotype? One might surmise that these cases represent an anatomically atypical form of AD or that they have concurrent neuropathologic processes masked by the AD. Previous work on a small number of cases showed that the anatomic distribution and the density of Alzheimer pathology in cases presenting clinically with PPA is not consistently different from cases presenting with dementia of the Alzheimer-type (DAT) [28]. Although these patients had a progressive aphasia that was initially unaccompanied by memory loss, the neurofibrillary tangle (NFT) counts were still many times higher in the hippocampo-entorhinal complex than in language cortices, and most of the patients showed no asymmetry of neocortical NFT even though they had phenotypically concordant symmetric atrophy of the left hemisphere. Another possibility that could resolve this apparent lack of clinico-anatomic concordance is the possibility of other pathologic processes. A limited survey in 11 cases did not reveal additional TDP-43 pathology in a group of 11 PPA-AD cases [28]. The current study represents an expansion of this investigation to include additional brain regions in a larger set of 53 cases.
Pathologic overlap of AD and tau-negative FTLD could not be ascertained prior to the identification of TAR DNA-binding protein of 43 kDa mw (TDP-43) as the major protein component in the ubiquitinated inclusions in the majority of cases of FTLD-U, now known as FTLD-TDP [3,7,9,36]. TAR DNA-binding protein (TDP-43) is normally located in the nucleus, and is functionally implicated in exon skipping and transcription regulation [3,7,9,36]. In most FTLD-U cases, neuronal cytoplasmic inclusions (NCIs), neuronal intranuclear inclusions (NIIs), and dystrophic neurites (DNs) are composed primarily of TDP-43, and normal nuclear TDP-43 is lost in neurons containing NCIs or NIIs. In most autopsy brains with AD pathology, tangles and plaques are so dense and widespread that they can mask tau- and ubiquitin-positive deposits characteristic of FTLD sub-types, so prior to the discovery of TDP-43, it was virtually impossible to identify pathologic overlap of AD and FTLD. Unless ubiquitin-positive neuronal intranuclear inclusions (NIIs) were identified in cases with pathologic AD, the diagnosis of concomitant FTLD-U could not be made with confidence. It remains difficult to diagnose concomitant FTLD-Tau, but TDP-43 IHC now provides a tool to immunohistochemically investigate the potential overlap of AD and FTLD-U/TDP, the most common FTLD sub-type [19, 22].
TDP-43-positive inclusions have been demonstrated in association with various other neurodegenerative disorders, including AD[1,4,17,18,21,39,41], hippocampal sclerosis (HS) [1, 41], dementia with Lewy bodies (DLB) [4, 17, 32], corticobasal degeneration (CBD) [41], Pick disease [3, 12], ALS-parkinson-dementia complex of Guam (ALS-PDC Guam) [14, 15, 29], argyrophilic grain disease (AGD) [13], Huntington disease (HD) [40], Perry syndrome [11, 44], and protein aggregate myopathies [37, 42]. They are also characteristic of sporadic amyotrophic lateral sclerosis (ALS) and familial ALS without SOD1 mutations [3, 23, 36]. In AD and HS, TDP-43 pathology is most often localized to the medial temporal regions-including the hippocampal dentate gyrus, entorhinal cortex, and amygdala (FTLD-TDP, limbic type), sometimes including involvement of superficial layers of the inferior temporal cortex. A minority of cases appear to have more widespread cortical involvement, so characteristic of FTLD-TDP, called FTLD-TDP, diffuse type by Amador-Ortiz et al. [1].
Because TDP-43 pathology may be present in up to 30% of AD cases [1,4,17,18,21,39,41], we reasoned that there might be an even greater proportion of cases with concomitant TDP-43 pathology in PPA and FTD syndromes with AD pathology in a way that may explain the clinical phenotype. In a previous study, TDP-43 immunostains of hippocampus, including dentate gyrus and entorhinal cortex, and frontal cortex were negative in 11 cases of PPA with AD pathology [28]. The current study looked in greater detail at the same 11, five additional cases of PPA, and 10 cases of FTD with AD pathology in order to determine whether concomitant FTLD-TDP pathology might be responsible for the clinical phenotypes. Additional regions included amygdala since TDP-43 pathology can be confined to this region, and inferior and superior temporal gyri. TDP-43 IHC was performed using three different immunostaining protocols on sections from the same anatomic regions and also on amygdala and inferior and superior temporal cortex from these 26 cases. After performing TDP-43 immunohistochemistry (IHC) on the PPA-AD and FTD-AD groups three times using different protocols and finding only a small percentage of cases with TDP-43 pathology, much less is reported in the literature for AD [1,4,17,18,21,39,41], we decided to perform TDP-43 IHC on a group of 27 cases of pathologic AD presenting with amnestic dementia (clinical diagnosis of DAT) for comparison.
Materials and methods
PPA and FTD groups
Of 60 Northwestern PPA and FTD brain autopsies (28 PPA, 32 FTD), 26 had the pathology of AD and 34 did not have pathologic AD; of these 34, 32 had FTLD without AD (21 FTLD-TDP, 9 FTLD-tau, and 2 FTLD-FUS), and two others had vascular dementia and non-specific morphologic changes. Average ages of onset and of death and average duration of disease were greater in the group with pathologic AD, reaching statistical significance in age of death (p = 0.005) and duration (p = 0.004) (see Table 1)。 The focus of this study was those cases of PPA (n = 16) and FTD (n = 10) with the pathology of AD. The cases with non-AD pathologic diagnoses are not part of the current study.
[align=center][align=center] 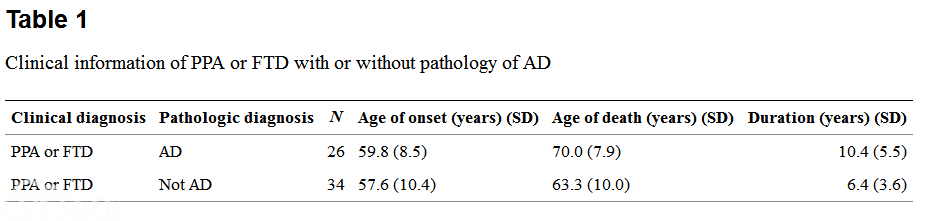 [/align][/align] DAT group
TDP-43 pathology in the PPA and FTD cases was compared to TDP-43 pathology in 27 cases of AD presenting clinically with DAT, for a total of 53 cases. Cases with concomitant Lewy body dementia (DLB) pathology were excluded from this group to avoid multiple variables.
Clinical information
Detailed clinical data were available from 14 of the 16 PPA and all of the 10 FTD patients with AD pathology. The diagnosis of primary progressive aphasia (PPA) was based on the performance on neuropsychological tests, a comprehensive clinical evaluation, and the criteria outlined by Mesulam [27]. The diagnosis of behavioral variant FTD (bvFTD) was made by a neurologist, performing a comprehensive clinical evaluation and neuropsychological testing, utilizing criteria outlined by the most recent work-group consensus on frontotemporal lobar degeneration [33]. The presence of changes in social-interpersonal behaviors, personality, and customary ways of behaving were paramount in this diagnostic category. The other FTD subjects had a variety of clinical deficits associated with frontotemporal function, including executive and attentional dysfunction in the absence of comportmental symptoms, or a mixture of behavioral and aphasic disturbances. Detailed clinical information was available from all of the 27 amnestic dementia patients. The diagnosis of probable AD was made according to currently accepted criteria [26].
Table 2 shows the clinical, pathologic, and genetic information in the study groups with pathologic AD. The average age of onset was significantly younger in the PPA-AD (p = 0.03) and FTD-AD (p = 0.0008) groups than in the probable AD/DAT-AD group. The average age of death in the PPA-AD group was younger than the DAT-AD group (p = 0.014) and older than the FTD-AD group (p = 0.048)。 The average age of death in the FTD-AD group was also younger than the DAT-AD group (p = 0.0003)。 Duration was shorter in the FTD-AD group than in the PPA-AD and DAT-AD groups, but the difference was not statistically significant. There was a higher male:female ratio in the PPA-AD and FTD-AD groups, while the opposite was true in the DAT-AD group, but the differences were not significant. The average brain weight in the FTD-AD group was significantly heavier than both the PPA-AD (p = 0.003) and DAT-AD (p = 0.007) groups, which may be related to the shorter duration for this group. The ApoE ε4 allele was present in 79% of the DAT-AD subjects, significantly higher than its presence in 36% of the PPA-AD subjects (p = 0.047) but similar to the FTD-AD group (78%) (p = 0.999)。 PROGRANULIN (PGRN) mutation analysis was performed on 10 of the 16 PPA and 7 of the 10 FTD cases, and no mutations were identified [5, 8]. This analysis included two of the three cases with TDP-43 pathology, one with NIIs and one without NIIs.
[align=center][align=center] 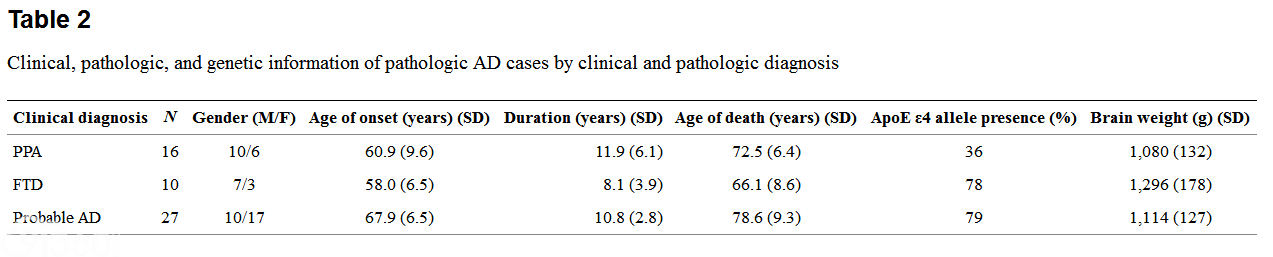 [/align][/align] Neuropathologic evaluation
Autopsies were performed by the Neuropathology Core of the Northwestern CNADC. All brains were evaluated grossly for cortical, caudate, hippocampal, cerebellar, and brainstem atrophy, and microscopically with H&E for neuronal loss, astrocytosis, superficial microvacuolation, and depopulation of pigmented nuclei. All brains were evaluated for AD pathology using thioflavine-S, Gallyas, tau IHC (AT8, Pierce-Endogen, Rockford, IL, USA), and A beta IHC (4G8, Signet, Dedham, MA, USA), for Lewy body pathology with α-synuclein (LB509, Zymed-Invitrogen, Carlsbad, CA, USA), and for non-Alzheimer- and non-Lewy-type deposits of tau, α-synuclein, and ubiquitin (DAKO polyclonal, Carpinteria, CA, USA)。
All of the cases with pathologic AD, regardless of the clinical diagnosis, had CERAD C, Braak V or VI, and NIA/Reagan high likelihood dementia due to AD [2, 6, 30]. Because we did not wish to exclude any of the PPA or FTD cases with AD pathology from this study, we did not exclude cases that had concomitant DLB. Two of the PPA cases also had DLB: one had DLB brainstem stage and one had DLB cortical stage or DLBD. One FTD case also had DLB limbic stage.
Ubiquitin immunohistochemistry
Briefly, 3-um sections from all cases were pre-treated with formic acid for 5 min and microwaved in 10 mM Citrate buffer pH 6.0 for 10 min, incubated with polyclonal ubiquitin antibody (Cat # Z0458, DAKO, Denmark), biotinylated secondary antibody, and Vectastain Elite ABC reagent (Vector Laboratories, Cat #PK 6100, Burlingame, CA, USA)。 Enhanced DAB metal substrate chromagen was used for visualization (Cat # 34065, Pierce-Endogen, Rockford, IL, USA)。 Slides were counterstained with hematoxylin for 30 s.
TDP-43 immunohistochemistry
Frontal (middle frontal gyrus, BA8-9) and temporal (superior temporal gyrus, BABA41-42) cortex, hippocampus with dentate gyrus, subiculum and parahippocampal gyrus, entorhinal cortex, and amygdala with inferior temporal gyrus (BA20) from all cases were immunostained with a polyclonal antibody to TDP-43 (Proteintech Group, Chicago, IL, USA)。 Four-micron sections were deparaffinized, pretreated in an antigen decloaking chamber at 100 °C with 10 mM citrate buffer, pH 6.0, for 20 min to enhance immunoreactivity, quenched for endogenous peroxidase, blocked for non-specific activity in 2% non-fat dried milk, and incubated with anti-TDP-43 antibody (1:1,000) overnight at 4°C. Sections were incubated with biotinylated goat anti-rabbit IgG secondary antibody (Biocare Medical, Concord, CA, USA) for 30 min followed by peroxidase-conjugated streptavidin in PBS with carrier protein (BioGenex, San Ramon, CA, USA) for 20 min and developed with AEC chromagen (Biogenex) for 5 min. Counterstaining was carried out with hematoxylin for 45 s and slides mounted with Advantage permanent aqueous mounting media (Innovex Biosciences)。
Because we did not see TDP-43 immunopositivity in 25-30% of our PPA or FTD cases with AD pathology, as had been published at that time [1], two additional TDP-43 immunohistochemical methods were used for the PPA or FTD with AD pathology cases, as outlined below. Following discussions with collaborators who used alternative protocols, TDP-43 IHC was repeated using a modified protocol. Sections were deparaffinized and quenched for endogenous peroxidase for 30 min in 300 ml of methanol containing 4 ml of commercially available stock 30% hydrogen peroxide. Antigen retrieval was performed with vector antigen unmasking solution (Vector laboratories, cat# H-330)。 Anti-TDP-43 antibody (Polyclonal, Proteintech, Chicago, IL, USA) at 1:8,000 in 0.1 M Tris/2% FBS was applied overnight at 4°C. The signal was developed using Biogenex Super Sensitive Polymer Detection kit (Biogenex QD430-XAK)。 Briefly, Super Enhancer was applied for 20 min at room temperature, followed by poly-HRP for 30 min and DAB chromagen (Biogenex, HK542-XAK) for 5 min, and counterstained with hematoxylin.
In addition, TDP-43 IHC was performed again at UT Southwestern on hippocampus/entorhinal cortex sections, following a previously published, enhanced, highly sensitive TDP-43 IHC protocol [16]. Briefly, for antigen retrieval, the sections were placed in a Pascal programmable pressure cooker (DakoCytomation, Carpinteria, CA, USA) containing Reveal solution (Biocare Medical, Concord, CA, USA), with the target temperature and time set to 126°C for 30 s. After the temperature dropped to 89°C, the slides were taken out and allowed to cool down at room temperature for 10 min. The primary antibody was a rabbit polyclonal antibody to TDP-43 at a dilution of 1:1,000 (ProteinTech, Chicago, IL, USA)。 The detection system was the alkaline phosphatase-based ultra View Red detection system (Ventana Medical Systems, Tucson, AZ, USA); the chromagen was Fast Red/Naphthol, and the procedure was performed using the Benchmark XT automated stainer (Ventana)。
TDP-43 pathology
TDP-43-labeled NCIs, NIIs, and DNs were estimated semiquantitatively; TDP-43 labeling of tangles and Lewy body pathology was noted but not included in this analysis. TDP-43-labeled neuronal cytoplasmic inclusions (NCIs), neuronal intranuclear inclusions (NIIs), and dystrophic neurites (DNs) in each region were semi-quantitatively estimated in three maximally labeled 200× fields and averaged. These were designated as “rare” (generally equivalent to <1 inclusion per 200× field), “sparse” (equivalent to 1-3 inclusions per 200× field), “moderate” (equivalent to 4-8 inclusions per 200× field), or “frequent” (equivalent to >9 inclusions per 200× field)。 For statistical analysis, these ratings were given numbers: rare 0.5, sparse 1, moderate 2, and frequent 3. Finally, for each case, the averages for all regions were added. The group score was then derived by dividing the added total scores by the number of cases in that group. This method of scoring allowed statistical comparisons between groups.
Confocal microscopy
Confocal microscopy was performed using previously described methods [10]. In brief, 6-μm sections were cut from formalin-fixed, paraffin-embedded tissues. Antigen retrieval was carried out using a decloaking chamber (Biocare Medical, Walnut Creek, CA, USA)。 Antibodies to TDP-43 and α-synuclein or TDP-43 and Tau (AT8) were used in combination as primary antibodies. Fluorescence signals were detected with donkey anti-rabbit IgG conjugated with FITC (Thermo Scientific, Rockford, IL, USA) and goat anti-mouse IgG conjugated with rhodamine (R-6393, Invitrogen, Carlsbad, CA, USA) using an LSM 510 META Laser Scanning Confocal Microscope with the multitracking setting. The same pinhole diameter was used to acquire each channel.
Hippocampal sclerosis
For both the hippocampal CA1 region and the subiculum, each case was semi-quantitatively estimated for neuronal loss and gliosis, from absent to severe, on a 0-3 point scale (0 not present; 1 mild; 2 moderate; 3 severe)。 Hippocampal sclerosis (HS) was defined as severe neuronal loss and gliosis in both regions.
Statistical analysis
Diagnostic groups were compared using one-way analysis of variance followed by post-hoc pairwise t tests for age, duration, and brain weight. Frequencies of gender and ApoE status were compared across groups using Fisher's exact test, which was also used for the post-hoc pairwise comparisons. The occurrence of positive TDP pathology (yes, no) and the ordinal scores for HS were compared across groups using Fisher's exact test. In the DAT-AD group, age was compared between positive and negative TDP groups using the independent sample t test. Age of onset and age of death were compared between TDP pathology groups (positive, negative) using the independent sample t test. Spearman correlation was used to relate TDP pathology scores with HS scores. Logistic regression analysis was used to relate TDP score (negative, positive) to HS scores adjusting for age of death. Statistical significance was indicated when p<0.05, and no adjustment for multiple testing was made.
Results
The modified TDP-43 IHC protocol, the UT Southwestern protocol, and the original TDP-43 IHC protocol gave similar results. In the PPA-AD and FTD-AD groups, there were three cases with TDP-43-positive inclusions of the type associated with FTLD-TDP. Case 26, an FTD-AD case with concomitant FTLD-U (based on the presence of ubiquitin-positive NIIs in the dentate gyrus), clearly also showed moderate numbers of NCIs and DNs and more frequent NIIs with TDP-43 IHC, and also had inclusions in the hippocampus, subiculum, and amygdala (Fig. 1), consistent with amygdala limbic TDP-43 pathology. This pathology had of course been masked by the AD pathology when ubiquitin IHC was used. An additional FTD-AD case, case 17, was found to have TDP-43-positive inclusions in frontal and temporal cortex, hippocampus, dentate gyrus, entorhinal cortex, and amygdala, consistent with diffuse (amygdala limbic neocortical) pathology, and this case was therefore re-classified as AD FTLD-TDP (Fig. 2)。 Case 16, a PPA-AD case with concomitant diffuse Lewy body disease (DLBD), had rare to sparse TDP-43-positive inclusions in the dentate gyrus, entorhinal cortex, and parahippocampal gyrus, and moderate inclusions in the amygdala, consistent with amygdala limbic stage. Rare amygdala inclusions had the morphology of Lewy bodies or neurofibrillary tangles (Fig. 3)。 TDP-43 pathology was not present in the other PPA case with DLB (brainstem stage) or the FTD case with DLB limbic stage. Except for these three cases, there was no TDP-43 immunopositivity of NCIs, DNs, or NIIs in any of the other PPA-AD or FTD-AD cases. All three cases had severe neuronal loss and gliosis in the hippocampal CA1 region and subiculum consistent with HS, as shown in Fig. 2 for case 17, and none of the other PPA-AD or FTD-AD cases had HS. In two additional cases, TDP-43 labeled amygdala inclusions with the morphologic appearance of tangles (not shown)。
[align=center][align=center]  [/align][/align]
[align=center][align=center]  [/align][/align] FTD case 17 has hippocampal sclerosis and TDP-43 immunoreactivity. a Severe neuronal loss and gliosis of both hippocampal CA1 region and subiculum consistent with hippocampal sclerosis. H&E stain, original magnification 20×。 b Dentate gyrus with TDP-43-positive NCI (long arrow), NII (arrowhead), and DNs (short arrows)。 Original magnification 600×。 c Frontal cortex with TDP-43-positive NCI (long arrow), NII (arrowhead), and DN (short arrow)。 Original magnification 400×.
[align=center][align=center] 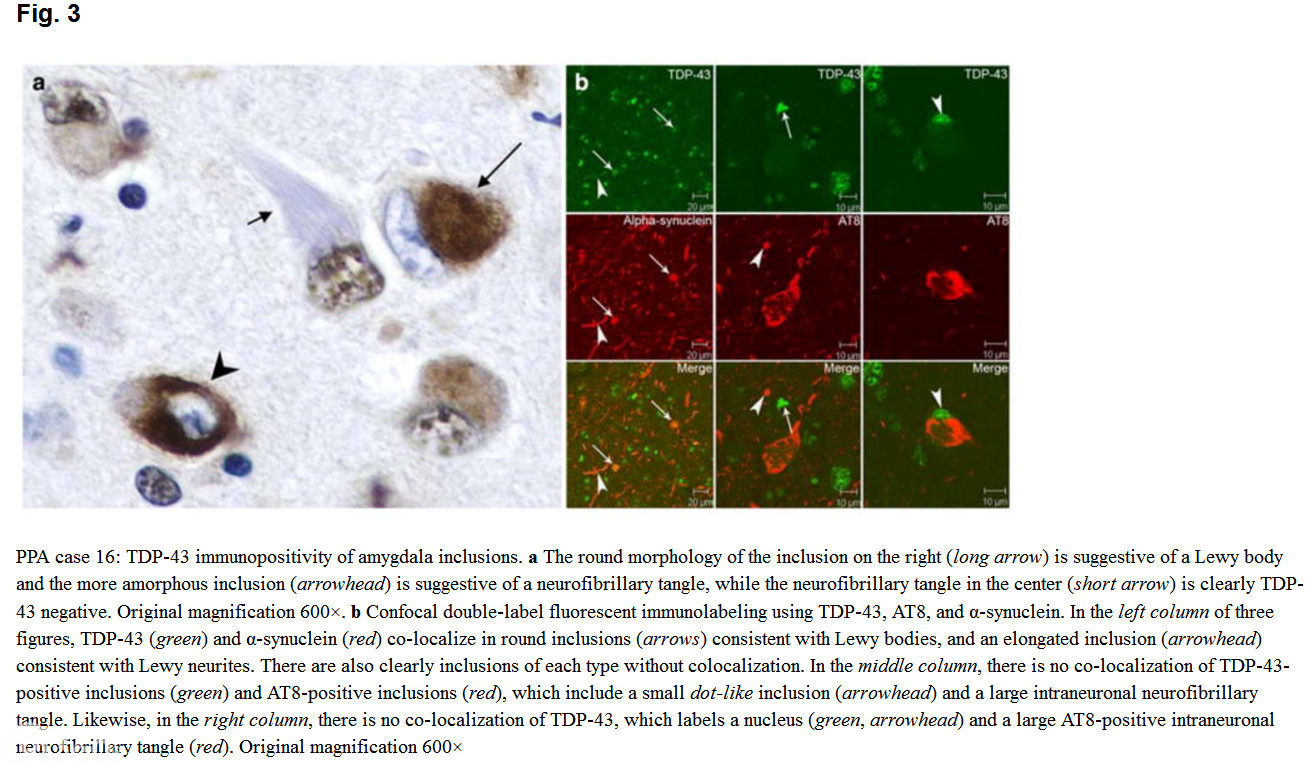 [/align][/align] The average TDP score for the combined PPA-AD/FTD-AD group was 0.6 (3/26 had positive TDP pathology)。 Taken separately, scores were 0.3 for the PPA group (1/16 or 6% positive TDP) and 1.1 for the FTD group (2/10 or 20% positive TDP)。
In the group of 27 DAT-AD cases, 14 cases (52%) had TDP-43 immunopositive inclusions. In ten cases, these were in dentate gyrus, amygdala, entorhinal cortex, or inferior temporal cortex (amygdala limbic stage)。 Four also had rare-moderate inclusions in either frontal or superior temporal cortex, consistent with diffuse TDP-43 pathology (inclusions in case 29 shown in Fig. 4)。 Two additional cases had TDP-43 immunopositivity of inclusions with the morphology of tangles.
[align=center][align=center] 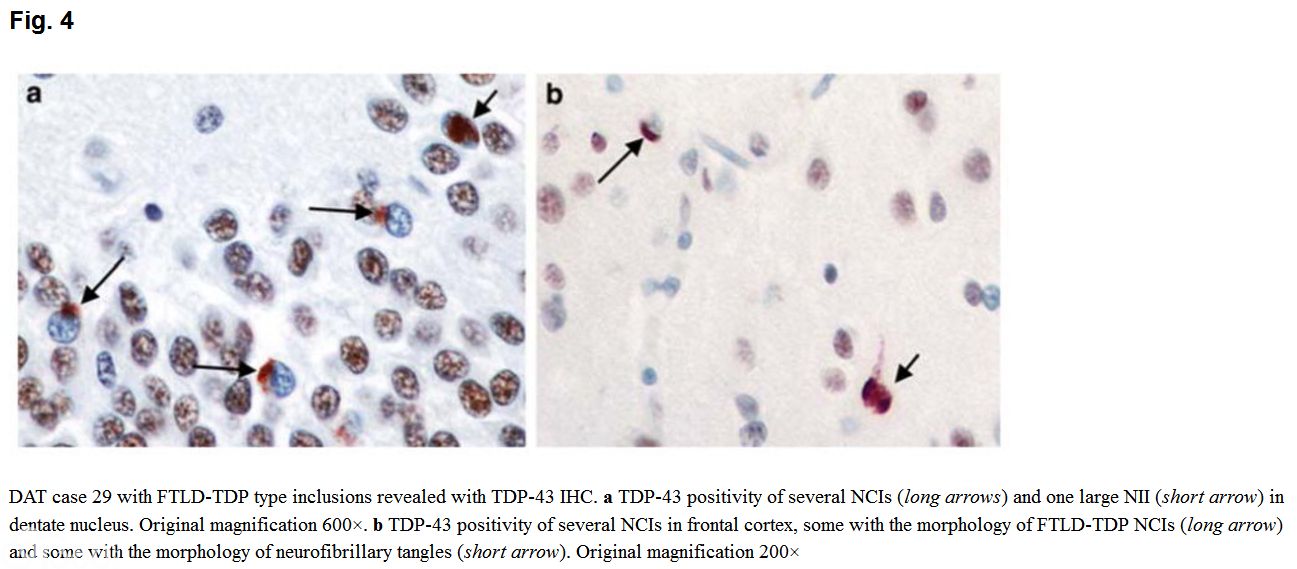 [/align][/align] The average TDP score for the DAT-AD cases was 3.5. In this group, TDP-43 pathology did not correlate with ApoE status. However, those with TDP-43 immunoreactivity were older at onset (p = 0.003) and death (p = 0.0003)。 Of the 14 cases with TDP-43 immunopositivity, 9 had HS. The average TDP score of these nine cases was 8.5.
Because TDP-43 pathology was found in only three of the 26 PPA-AD and FTD-AD cases (12%), which is much less than the previously reported [1,4,17,18,21,39,41], but was found in 52% of the DAT-AD cases, higher than the previously reported, we analyzed our data to determine why this was so. Paucity of TDP-43 pathology in the PPA-AD and FTD-AD groups precluded a correlation between TDP-43 and neuronal loss/gliosis within each of these groups. For all three groups combined, the occurrence of positive TDP pathology (yes, no) was highly correlated with both hippocampal and subicular neuronal loss/gliosis (Fisher's exact test)。 In the DAT-AD group, TDP pathology score correlated with both hippocampal (p<0.0001) and subicular (p<0.0001) neuronal loss and gliosis (Spearman correlation)。 The occurrence of TDP pathology in the DAT-AD (52%) was significantly greater than in the PPA-AD group (6%, p = 0.003) but not significantly greater than the FTD-AD group (20%, p = 0.14)。 Hippocampal (p = 0.003) and subicular (p = 0.009) neuronal loss and gliosis were significantly greater in the DAT-AD than in the PPA-AD group. Multivariate logistic regression adjusting for age of death was performed in all the cases with TDP-43 pathology, regardless of clinical phenotype. This analysis demonstrated that subicular neuronal loss and gliosis was the only pathologic predictor of TDP-43 immunoreactivity (p = 0.002)。
Table 3 shows the semiquantitative estimates for regional TDP-43 pathology and hippocampal and subicular neuronal loss and gliosis for each case. Table 4 summarizes the average TDP scores for each group and the TDP-43 regional sub-types in each group.
[align=center][align=center] 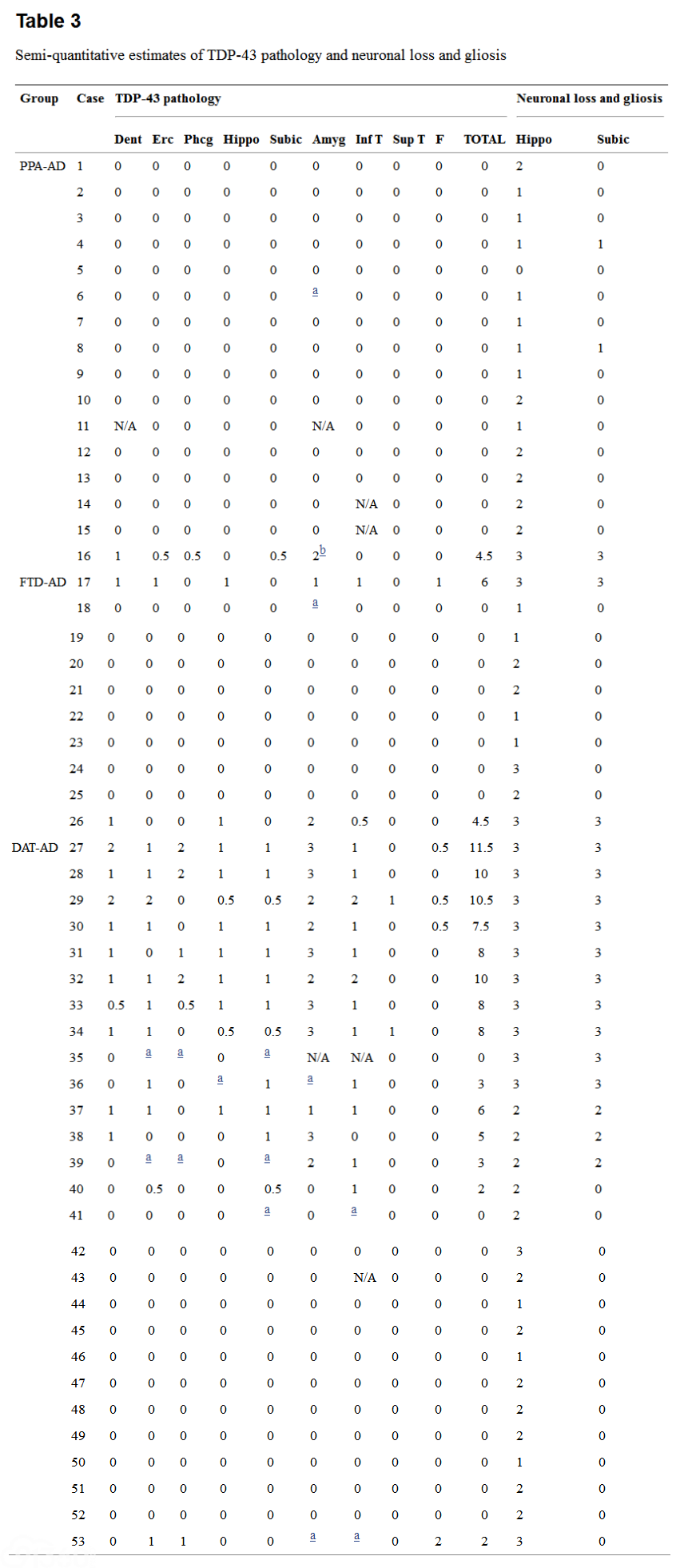 [/align][/align] For TDP-43 pathology, 0.5 rare, 1 sparse, 2 moderate, 3 frequent, N/A not available
For neuronal loss and gliosis, 0 not present, 1 mild; 2 moderate, 3 severe
Dent dentate gyrus, Erc entorhinal cortex, Phcg parahippocampal gyrus, Hippo hippocampus, Subic subiculum, Amyg amygdala, Inf T inferior temporal gyrus, Sup T superior temporal gyrus, F frontal cortex
aNeurofibrillary tangles only
bFTLD-TDP pathology and Lewy bodies
[align=center][align=center] 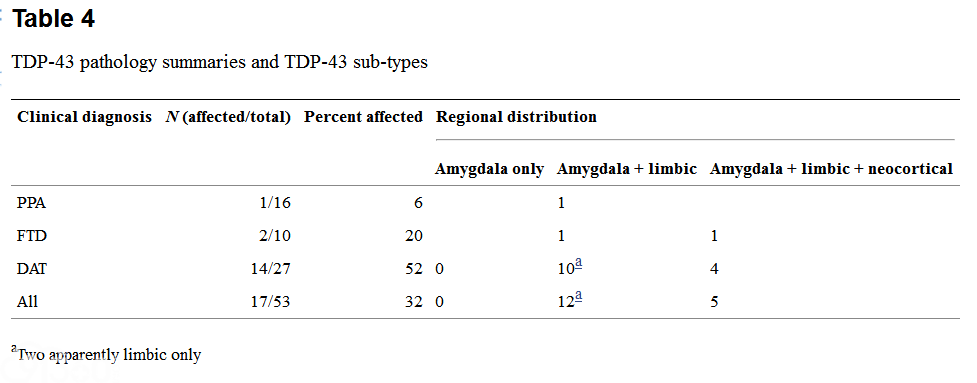 [/align][/align] Discussion
From the neuropathologist's point of view, AD is defined by the histologic presence of NFTs and neuritic plaques (NPs) in a characteristic density and distribution [2, 6, 30]. From the clinician's perspective, DAT, the most typical clinical form of pathologic AD, is characterized by a progressive amnestic dementia with memory loss the most prominent early sign [27, 28]. There is general concordance of these clinical and pathologic diagnoses: up to 90% of clinically diagnosed late-onset DAT cases have AD pathology at autopsy. This relationship fits the known early predilection of AD morphologic pathology, NFTs and eventual neuronal loss, for the medial temporal parts of the brain known to control memory formation.
In addition to amnestic dementia, clinical syndromes of dementia include PPA and FTD. These clinical syndromes, especially the PPA variants, can leave memory intact, and would therefore be expected to have a different neuropathologic basis than typical AD. In fact, most cases of clinically diagnosed PPA and FTD have neuropathologic changes belonging to the family of frontotemporal lobar degenerations (FTLD)。 These include FTLD-TDP with or without ALS pathology, FTLD-UPS (most of which are now FTLD-FUS), FTLD-tau [including Pick disease, corticobasal degeneration (CBD), progressive supranuclear palsy (PSP), and “unclassifiable tauopathy” pathologic phenotypes], DLDH, and AD pathology [24, 27, 28, 34]. FTLD-TDP most often presents clinically with either FTD or PPA, and many patients with FTLD-TDP have clinical and pathologic ALS.
In keeping with the clinical syndrome, the neuronal loss may be most severe in the frontal lobes for FTD and the left perisylvian cortex for PPA. In our experience, PPA cases with pathologic AD also show asymmetric left perisylvian atrophy but apparently without corresponding concentration of plaques and tangles. There is one report of the presence of argyrophilic thorny astrocyte clusters (ATAC) in seven of eight cases of PPA-AD and none of six cases of DAT-AD [31]. We previously reported finding multiple clusters of ATAC in only 2 of 11 cases of PPA-AD [28]. In the current study, we found multiple ATAC in one additional FTD-AD and no additional PPA-AD cases. We also found multiple ATAC in two DAT-AD cases and sparse ATAC in two others. Thus far, then, we re-affirm our previous conclusion that these clusters of ATAC may be interesting to follow up on, but they do not appear to provide an answer that is applicable to all PPA-AD cases [28].
The PPA-AD and FTD-AD cases in the current study had significantly older ages at death and duration of disease than the PPA and FTD cases with non-AD pathology (Table 1), and we considered the possibility that their increased age made it more likely for AD pathology to be masking an underlying pathologic FTLD. TDP-43 immunohistochemistry allows one, in most cases, to distinguish between FTLD-TDP and AD, but we found TDP-43 pathology characteristic of FTLD-TDP in only three of the 26 cases (12%) of PPA-AD or FTD-AD.
The difference in the prevalence of TDP-43 pathology in our PPA-AD and FTD-AD groups (12%) from what has been reported in AD in general (30%) [1,4,17,18,21,39,41] is striking, and significant (p = 0.046)。 Although it confirms the findings in our initial study [28], and is similar to a report by Josephs et al. [20] of PPA with AD pathology, it is very different from the 30% reported by Amador-Ortiz et al. in phenotypically typical AD and subsequently confirmed by others [1,4,17,18,21,39,41]. In the group of 26 PPA-AD or FTD-AD cases in the current paper, one could reasonably expect to find TDP-43-positive inclusions in seven or eight cases if the 30% incidence of TDP-43 abnormality is a general feature of AD pathology. Instead, we found “diffuse type TDP-43 immunoreactivity” in only one case, i.e., sufficient cortical and subcortical TDP-43 immunoreactivity for the pathologic diagnosis of FTLD-TDP, and limbic stage TDP-43 pathology in two. One of these, and two additional cases in this group, had TDP-43 labeling of a small subset of medial temporal neurofibrillary tangles or Lewy bodies/neurites. The significance of combined immunopositivity for tau or α-synuclein and TDP-43 is not completely clear. However, it raises the possibility that TDP-43 may contribute to pathogenesis in a subset of PPA. There is one report that the presence of TDP-43 immunoreactivity in AD modifies the clinicopathologic phenotype in AD [21]. It is unclear whether this is the case in the current study. TDP-43 immunoreactivity of any type was seen in only two PPA and three FTD cases, making clinicopathologic correlation difficult. Ultimately, though, in the group of PPA and FTD cases with pathologic AD, the incidence of concomitant TDP-43 pathology was less than that found in unselected cases of pathologic AD [1].
What is the basis for this difference? One study showed that age of death correlated with TDP-43 pathology in 14 cases of AD, one of two series reported; the other series, containing 19 cases, showed no correlation [3]. Another study showed that ages of both onset and death correlated with TDP-43 pathology, but multivariate logistic regression adjusting for age of death showed that TDP-43 pathology correlated only with hippocampal sclerosis [21]. It is interesting that both studies chose cases with the pathologic diagnosis of AD and neither compared TDP-43 immunoreactivity by clinical diagnosis. In the DAT-AD group, ages of onset and death correlated with TDP-43 pathology. When we looked at all three groups together, i.e., regardless of the clinical diagnosis, multivariate logistic regression adjusting for age of death revealed that subicular neuronal loss and gliosis was the only pathologic predictor of TDP-43 immunoreactivity (p = 0.003)。 This is similar to a previous report correlating TDP-43 pathology with HS [21]. Interestingly, in the current study, hippocampal neuronal loss and gliosis was not a pathologic predictor of TDP-43 immunoreactivity, suggesting that there is a molecular/pathoetiologic difference between HS with and without subicular neuronal loss. Also interesting is that when all three groups are combined, the percentage of cases with pathologic AD having TDP-43 pathology is 32%, close to the percentage previously reported in pathologic AD unselected for clinical diagnosis [1,4,17,18,21,39,41].
Similar to our finding that only one of the 16 PPA-AD cases had TDP-43 pathology and HS, another study of five PPA-AD patients showed that none had TDP-43 pathology [20]. HS was not specifically mentioned in this paper, but hippocampal neuronal loss and gliosis was less in this group than in those with “typical AD” [20]. Finally, an analysis of 14 pathologic AD cases presenting clinically with PPA (6 cases), bvFTD (3 cases), and corticobasal syndrome (CBS) (5 cases) demonstrated TDP-43 pathology in only one case, while in the comparison group of 14 DAT-AD case 29% had TDP-43 pathology [43]. Interestingly, concurring with the lack of memory impairment as a prominent clinical feature, none of the “atypical AD” cases had severe neuronal loss and gliosis in the hippocampus [43]. This differs somewhat from our PPA-AD and FTD-AD groups in that the three cases with TDP-43 pathology did have severe hippocampal and subicular neuronal loss and gliosis. This may be because of our larger sample size of 16 PPA and 10 FTD cases, or simply a coincidental fact.
In their paper, Amador-Ortiz and colleagues [1] stated that “most” of their AD cases with TDP-43 immunopositivity had concomitant HS. Because cases of HS alone are known to have TDP-43 immunopositive inclusions [1, 38], the question is whether AD cases with TDP-43 immunopositivity have concomitant full-blown FTLD-TDP or simply HS with TDP-43 proteinopathy [18]. Significantly, the only PPA-AD or FTD-AD cases that had TDP-43 pathology were those cases with concomitant HS. It is most interesting that in the DAT-AD group with TDP-43 pathology, the majority of cases had HS, and those with TDP-43 pathology without HS had less severe TDP-43 pathology. These cases also had much less neocortical and striatal pathology than the cases with FTLD-TDP. Do our DAT-AD cases with TDP-43 pathology have combined pathologic AD and FTLD-TDP, combined pathologic AD and HS with TDP-43 proteinopathy, or simply AD with TDP-43 proteinopathy? There is likely not one simple answer to this question, but additional studies investigating these relationships are in progress, which may, in addition, shed light on the question of what the minimal pathologic requirements are for the diagnosis of FTLD-TDP. In conclusion, this study shows that in the current series of clinical PPA and FTD, FTLD-TDP is rarely combined with pathologic AD, and that medial temporal TDP-43 pathology is more tightly linked to HS than to clinical phenotype. These findings challenge the current notions about clinicopathologic correlation, especially about the role of multiple pathologies.
Finally, could the cases in the current study have concomitant FTLD-FUS? The discovery that FUS appears to be the major protein component of the TDP-43-negative inclusions in FTLD-UPS/“atypical FTLD-U” [24, 35] exposes a new avenue for us to investigate the basis of the clinical syndrome of PPA or FTD in cases with pathologic AD.
|





 发表于 2017-12-20 18:30:55
发表于 2017-12-20 18:30:55

